

Contents
Rapid Communications
Ion beam modification of fullerene films and their frictional behavior
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1615-1618
-
- Article
- Export citation
Diamond nucleation by carbon fibers on unscratched substrate by hot-filament chemical vapor deposition
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1619-1621
-
- Article
- Export citation
Deposition of molybdenum carbonitride thin films from Mo(NBut)2(NHBut)2
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1622-1624
-
- Article
- Export citation
Articles
Superconductivity of epitaxially grown α-axis YBCO films on SrTiO3 substrate
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1625-1632
-
- Article
- Export citation
Formation and coarsening behavior of Y2BaCuO5 from peritectic decomposition of YBa2Cu3O7−x
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1633-1643
-
- Article
- Export citation
Surface crystallographic structure compatibility between substrates and high Tc (YBCO) thin films
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1644-1656
-
- Article
- Export citation
Effects of oxygen pressure on the structure of Y–Ba–Cu–O materials formed by containerless melting and solidification
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1657-1660
-
- Article
- Export citation
Effects of oxygen partial pressure on the crystallization of amorphous Bi–Sr–Ca–Cu–O and Bi–Sr–Ca–Cu–O + Ag
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1661-1671
-
- Article
- Export citation
A systematic study of the synthesis of useful Tl2Ba2Ca2Cu3O10−δ bulk superconductor
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1672-1677
-
- Article
- Export citation
High quality a-Si/Nb and a-SiN/NbN artificial multilayers for Josephson applications
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1678-1682
-
- Article
- Export citation
Ultralow load indentation hardness and modulus of K – and α–Al2O3 CVD coatings
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1683-1692
-
- Article
- Export citation
Identification of fracture sequences during sharp indentation of polycrystalline Al2O3
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1693-1700
-
- Article
- Export citation
Reactive melt infiltration of silicon-niobium alloys in microporous carbons
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1701-1708
-
- Article
- Export citation
-
Studies of the reactive melt infiltration of silicon-niobium alloys in microporous carbon preforms prepared by the pyrolysis of a polymer precursor have been carried out using modeling, DTA, and melt infiltration. Mercury porosimetry results indicate a very narrow pore size distribution with virtually all the porosity within the carbon preforms open to infiltrants. The morphology and amount of the residual phases (niobium disilicide and silicon) in the infiltrated material can be tailored according to requirements by careful control of the properties (pore size and pore volume) of the porous carbon preforms and alloy composition. The average room temperature four-point fiexural strength of a reaction-formed silicon carbide material (made by the infiltration of medium pore size carbon preform with Si–5 at. % Nb alloy) is 290 ± 40 MPa (42 ± 6 ksi) and the fracture toughness is 3.7 ± 0.3
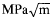 . The fiexural strength decreases at high temperatures due to relaxation of residual thermal stresses and the presence of free silicon in the material.
. The fiexural strength decreases at high temperatures due to relaxation of residual thermal stresses and the presence of free silicon in the material.
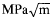 . The fiexural strength decreases at high temperatures due to relaxation of residual thermal stresses and the presence of free silicon in the material.
. The fiexural strength decreases at high temperatures due to relaxation of residual thermal stresses and the presence of free silicon in the material.Porous alumina ceramics by spray-pyrolyzed powder from aluminum sulfate and aluminum nitrate solutions
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1709-1713
-
- Article
- Export citation
A 27Al MAS nmr study of Synroc crystallization from alkoxide precursors
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1714-1720
-
- Article
- Export citation
Metal-organic chemical vapor deposition of ZrO2 films using Zr(thd)4 as precursors
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1721-1727
-
- Article
- Export citation
Formation kinetics of PbZrxTi1−xO3 thin films
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1728-1733
-
- Article
- Export citation
A new procedure for measuring the decohesion energy for thin ductile films on substrates
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1734-1741
-
- Article
- Export citation
Effects of boron on the deformation behavior of Ni3Al
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1742-1754
-
- Article
- Export citation
Effects of ternary additions on the twin energy and site preference in γ-TiAl
-
- Published online by Cambridge University Press:
- 03 March 2011, pp. 1755-1760
-
- Article
- Export citation