For a population undergoing mass selection, derived from an unselected base population in generation zero, the expected long-term contribution to the population of an ancestor from generation 1 was shown to be equal to that expected during random selection multiplied by 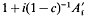 (where
(where 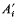 is one half the breeding value of the ancestor for the trait under selection standardized by the phenotypic standard deviation, i the intensity of selection, and
is one half the breeding value of the ancestor for the trait under selection standardized by the phenotypic standard deviation, i the intensity of selection, and 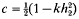 is the competitiveness which is defined by
is the competitiveness which is defined by 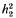 the heritability in generation 2 and k the variance reduction coefficient). It was shown that the rate of inbreeding (ΔF) could be partitioned into three components arising from expected contributions, sampling errors and sampling covariances respectively. Using this result ΔF was derived and shown to be dominated by terms that describe ΔF by variance of family size in a single generation plus a term that accounts for the expected proliferation of lines over generations from superior ancestors in generation 1. The basic prediction of ΔF was given by
the heritability in generation 2 and k the variance reduction coefficient). It was shown that the rate of inbreeding (ΔF) could be partitioned into three components arising from expected contributions, sampling errors and sampling covariances respectively. Using this result ΔF was derived and shown to be dominated by terms that describe ΔF by variance of family size in a single generation plus a term that accounts for the expected proliferation of lines over generations from superior ancestors in generation 1. The basic prediction of ΔF was given by
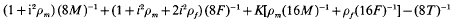
where M and F are the numbers of breeding males and females, T the number of offspring of each sex, ρm and ρt are correlations among half-sibs in generation 2 for males and females respectively, and K is a function of the intensity and competitiveness.
