
3 results
On a wider class of prior distributions for graphical models
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 08 June 2023, pp. 230-243
- Print publication:
- March 2024
-
- Article
-
- You have access
- HTML
- Export citation
-
Gaussian graphical models are useful tools for conditional independence structure inference of multivariate random variables. Unfortunately, Bayesian inference of latent graph structures is challenging due to exponential growth of
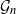 $\mathcal{G}_n$, the set of all graphs in n vertices. One approach that has been proposed to tackle this problem is to limit search to subsets of
$\mathcal{G}_n$, the set of all graphs in n vertices. One approach that has been proposed to tackle this problem is to limit search to subsets of 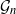 $\mathcal{G}_n$. In this paper we study subsets that are vector subspaces with the cycle space
$\mathcal{G}_n$. In this paper we study subsets that are vector subspaces with the cycle space 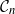 $\mathcal{C}_n$ as the main example. We propose a novel prior on
$\mathcal{C}_n$ as the main example. We propose a novel prior on 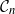 $\mathcal{C}_n$ based on linear combinations of cycle basis elements and present its theoretical properties. Using this prior, we implement a Markov chain Monte Carlo algorithm, and show that (i) posterior edge inclusion estimates computed with our technique are comparable to estimates from the standard technique despite searching a smaller graph space, and (ii) the vector space perspective enables straightforward implementation of MCMC algorithms.
$\mathcal{C}_n$ based on linear combinations of cycle basis elements and present its theoretical properties. Using this prior, we implement a Markov chain Monte Carlo algorithm, and show that (i) posterior edge inclusion estimates computed with our technique are comparable to estimates from the standard technique despite searching a smaller graph space, and (ii) the vector space perspective enables straightforward implementation of MCMC algorithms.
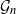
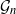
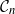
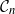
Identifiability of stochastically modelled reaction networks
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 32 / Issue 5 / October 2021
- Published online by Cambridge University Press:
- 15 February 2021, pp. 865-887
-
- Article
- Export citation
Networks' characteristics are important for systems biology
-
- Journal:
- Network Science / Volume 2 / Issue 2 / August 2014
- Published online by Cambridge University Press:
- 03 September 2014, pp. 139-161
-
- Article
- Export citation
