



Introduction
Parkinson’s disease (PD) is the second most common of the neurodegenerative disorders(Reference Hirtz, Thurman and Gwinn-Hardy1,Reference Wirdefeldt, Adami and Cole2) . It affects approximately 1 % of individuals over the age of 60 years worldwide(Reference Bray and Huggett3,Reference Tysnes and Storstein4) and continues to be a critical area of research focus. Symptoms and diagnoses typically appear later in life, at an average age in the range of 64–66 years(Reference Wong, Gilmour and Ramage-Morin5). Due to an increasingly ageing population(6), the number of those affected by PD is expected to grow substantially in coming years(Reference Bach, Ziegler and Deuschl7). Some estimates suggest an increase of 65 % for those over age 40 years by 2031(Reference Bray and Huggett3), and others for the amount of affected individuals to double by 2050(Reference Bach, Ziegler and Deuschl7).
PD is characterised by the progressive degeneration of dopaminergic (DA) neurons within the substantia nigra pars compacta (SNc) region of the midbrain. This degeneration appears to be specific, as mesolimbic DA neurons of the ventral tegmental area remain largely unaffected. The death of the SNc DA neuronal population results in a depletion of striatal dopamine, which in turn leads to a range of motor impairments. At the onset of symptoms, approximately 60 % of SNc DA neurons are generally believed to have already been lost(Reference Dauer and Przedborski8). As a result, individuals experience rigidity of movement, tremors at rest, and a slowing or absence of voluntary movement(Reference Dauer and Przedborski8). These impairments affect daily tasks such as walking, writing, dressing and eating, all of which contribute to an overall negative impact on quality of life. Using a measure incorporating eight functional categories (cognition, emotion, speech, vision, mobility, dexterity, pain and discomfort), individuals living with PD were found to have a significantly reduced quality of life, which translated to moderate to severe disability in 82 % of individuals(Reference Bray and Huggett3). Because PD is chronic and progressive, symptoms also deteriorate further over time, leading to greater detriments to quality of life for patients.
In addition to these effects, those with PD have shown significantly greater risk for developing dementia compared with controls(Reference Aarsland, Andersen and Larsen9,Reference Hobson and Meara10) . In PD dementia, diagnosis of dementia occurs after at least 1 year has passed since PD diagnosis. This form of dementia can be characterised by dysfunction in executive and visuospatial abilities, attention and memory, with the latter typically less affected than seen in Alzheimer’s disease(Reference Dubois, Burn and Goetz11,Reference Poewe, Gauthier and Aarsland12) . A link is also suspected between another type of dementia, known as Lewy body dementia, and PD. In this form, symptoms of dementia occur before or in conjunction with motor impairment(Reference Zupancic, Mahajan and Handa13). Cytoplasmic inclusions called Lewy bodies, consisting primarily of α-synuclein protein, can be found within brain tissue in PD resulting in Lewy body dementia, and may contribute to cognitive symptoms(Reference Baba, Nakajo and Tu14,Reference Spillantini, Crowther and Jakes15) . Though Lewy bodies are observed in other neurological conditions and are therefore not specific to PD, they are commonly found in patients with PD and are considered a pathological feature of the disease(Reference Spillantini, Crowther and Jakes15).
Research dedicated to PD continues to be of critical importance as the precise cause of the neurodegeneration affecting DA neurons remains unknown. As a result, current methods of treatment fail to target a cause, and instead treat only the identifiable symptoms. For instance, the widely used drug levodopa (l-DOPA (l-3,4-dihydroxyphenylalanine)) helps replenish lost striatal dopamine but fails to address the underlying cell death occurring within the SNc. Despite its ubiquitous use in the treatment of PD, l-DOPA can also have limited effectiveness, wear off over time, and elicit undesirable side effects(Reference Stacy, Bowron and Guttman16). As such, the need for further research to develop more effective and targeted treatments for those with PD is imperative.
Genetic and environmental factors have been the topic of numerous investigations, and have provided some insight into potential pathological mechanisms, such as α-synuclein aggregation and increased oxidative stress. Recently, emerging evidence has indicated a potential role for dietary factors(Reference Seidl, Santiago and Bilyk17), particularly B-vitamins, in the development of PD. Folate, a B-vitamin, is found naturally in foods such as leafy green vegetables and liver. A synthetic form of folate, folic acid, is found within vitamin supplements and has proven ability as a preventative measure against neural tube defects(Reference Ray, Meier and Vermeulen18). The protective effects of folates in neural tube defects led to mandatory fortification with folic acid in a number of countries worldwide(Reference Crider, Bailey and Berry19). In addition to the protective effects on neural tube development, folates are involved in nucleotide, protein and neurotransmitter synthesis, DNA repair, methylation, second messenger systems, ion channels, as well as the metabolism of homocysteine in the brain(Reference Murray, Emmerson, Jadavji and Lee20).
One-carbon metabolism is outlined in Fig. 1. Within cells, folate is converted to its main circulating form, 5-methyltetrahydrofolate (5-methylTHF), by the enzyme methylenetetrahydrofolate reductase (MTHFR). 5-MethylTHF is used by methionine synthase and vitamin B12 to remethylate homocysteine into methionine. The enzyme methionine synthase reductase (MTRR) maintains adequate levels of cobalamin for methionine synthase. Methionine can be converted to the methyl donor S-adenosylmethionine (SAM). After losing its methyl group, SAM becomes S-adenosylhomocysteine (SAH), which can then be metabolised further into homocysteine. Folate is therefore a cofactor in the metabolism of homocysteine, through its conversion to 5-methylTHF, which allows for remethylation of homocysteine into methionine.
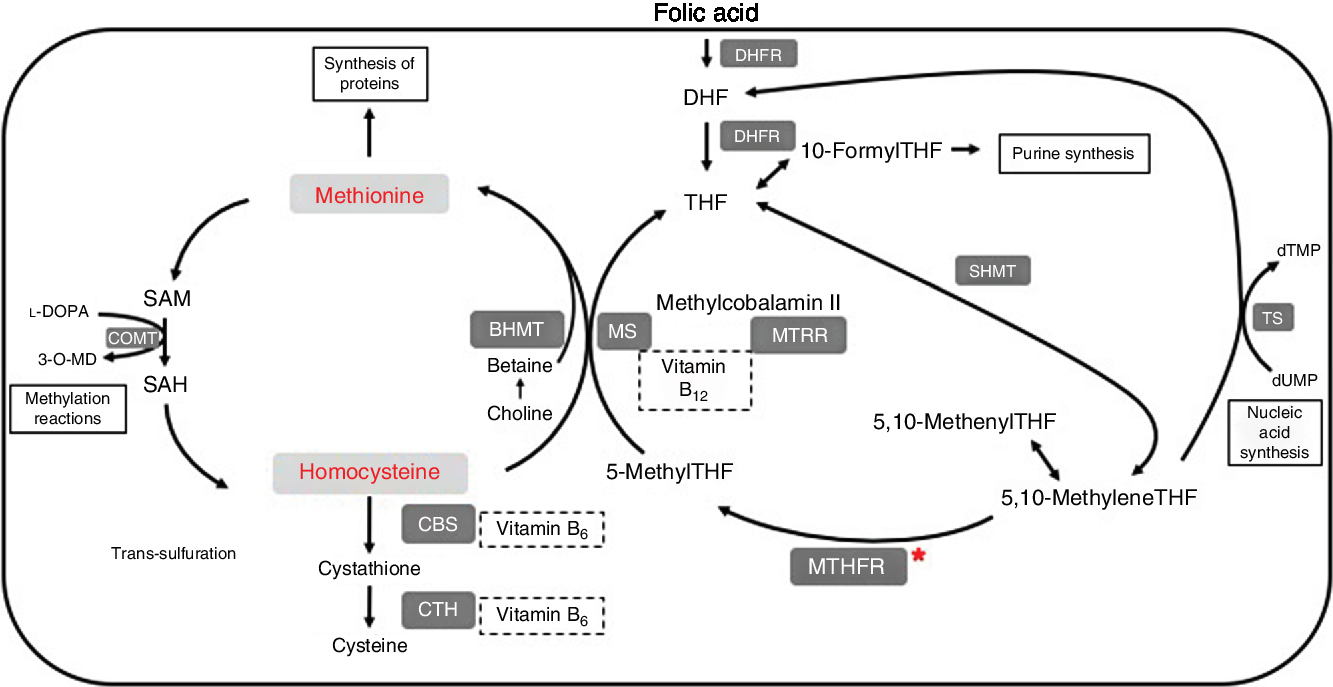
Fig. 1. Summary of one-carbon metabolism and levodopa (l-DOPA or l-3,4-dihydroxyphenylalanine) interaction. 3-O-MD, 3-O-methyldopa; BHMT, betaine homocysteine methyltransferase; CBS, cystathionine-β-synthase; COMT, catechol-O-methyltransferase; CTH, cystathionine γ-lyase; DHF, dihydrofolate; DHFR, dihydrofolate reductase; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; MS, methionine synthase; MTHFR, methylenetetrahydrofolate reductase; MTRR, methionine synthase reductase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate; TS, thymidylate synthase.
Elevations in homocysteine can occur through several processes. Levels of folate and homocysteine generally show an inverse relationship, with low amounts of folate associated with elevations in homocysteine and vice versa. As a cofactor in the metabolism of homocysteine, low levels of folate result in a reduction in the amount of 5-methylTHF. Lower levels of 5-methylTHF mean a decline in the amount of homocysteine undergoing reconversion to methionine, allowing homocysteine to accumulate as a result. Therefore, a failure to obtain adequate levels of folates within the diet can be associated with high concentrations of homocysteine. Similarly, low dietary intake of vitamins B6 and B12 also leads to increased levels of homocysteine.
Trans-sulfuration is an alternative pathway to reduce levels of homocysteine. In this reaction, cystathionine-β-synthase (CBS) converts homocysteine to cystathionine. The CBS knockout mouse model, the CBS –/– mouse, has significantly elevated plasma homocysteine levels (about 200 µm) and a low survival rate(Reference Watanabe, Osada and Aratani21,Reference Specific, Choumenkovitch and Selhub22) . The mice that do survive do not develop neuropathologies(Reference Watanabe, Osada and Aratani21,Reference Troen23) . This may be because CBS –/– mice do not have DNA methylation inhibition in brain tissue(Reference Specific, Choumenkovitch and Selhub22).
Alternatively, homocysteine levels can become elevated due to reduced levels of enzymes within one-carbon metabolism. For example, this can occur as a result of mutations in genes encoding for enzymes such as MTHFR or MTRR(Reference Gorgone, Currò and Ferlazzo24,Reference Yuan, Sheu and Yu25) . A polymorphism in MTHFR 677C→T leads to increased levels of homocysteine(Reference Frosst, Blom and Milos26). MTHFR-deficient mice have increased levels of homocysteine when compared with wild types(Reference Chen, Karaplis and Ackerman27). Reduced levels of MTRR increase homocysteine in humans(Reference Zavadakova, Fowler and Zeman28). A gene trap mouse model of MTRR has mildly increased levels of homocysteine(Reference Elmore, Wu and Leclerc29,Reference Jadavji, Bahous and Deng30) .
Increased levels of homocysteine have been associated with the development of several pathological conditions, including PD(Reference Rodriguez-Oroz, Lage and Sanchez-Mut31). Interestingly, patients with PD have consistently been shown to have elevated levels of homocysteine compared with controls(Reference Rodriguez-Oroz, Lage and Sanchez-Mut31–Reference Paul and Borah36). Furthermore, levels of homocysteine are increased in PD patients administered l-DOPA(Reference Wurtman37,Reference Moskowitz and Wurtman38) , as l-DOPA can be methylated using methyl groups from SAM, resulting in increases in SAH and homocysteine. Previous work has demonstrated that elevated levels of homocysteine increase risk for vascular disease(Reference Castro, Rivera and Blom39). PD patients with elevated levels of homocysteine may develop vascular disease(Reference Kuhn, Roebroek and Blom40,Reference Müller, Werne and Fowler41) .
At present, it is unknown how precisely nutritional factors might play a role in PD. Given the close relationship between B-vitamins and homocysteine, the latter of which has shown an association with PD, it seems likely that these dietary factors have at least some level of involvement. The importance of understanding all factors, both genetic and environmental, at play in the degenerative process is clear in order to develop more effective and targeted treatments for those with PD. Such knowledge could also lead to significant improvements in quality of life for those affected by PD. With this in mind, and with emerging evidence that has indicated a possible role for dietary factors in PD development(Reference Seidl, Santiago and Bilyk17), investigations into nutritional factors appear particularly timely since therapeutic development for PD has been slow. The aim of the present review is to determine whether one-carbon metabolism and homocysteine are involved in PD onset and related pathology and identify potential mechanisms related to this.
Materials and methods
Publications using medical subject headings (MeSH) keywords, one-carbon metabolism, folate, folic acid, MTHFR, Parkinson’s disease, l-DOPA and homocysteine were retrieved. Studies involving human subjects were categorised into three areas: polymorphisms in one-carbon metabolism; dietary intake of B-vitamins; and methylation. For each study we collected the following data: the country in which the study was conducted; whether government fortification was present; study design; sample size; and the main findings of the study. All of these findings are summarised in Tables 1 to 4. Case–control, prospective cohort, meta-analysis and non-randomised trials were included in the review. To understand the mechanistic interactions of one-carbon metabolism on PD pathology, we analysed in vitro and in vivo studies. For each study, we collected the model system information, design of the study, and the major findings. These results are summarised in Table 5.
Table 1. Impact of polymorphisms of enzymes in folate metabolism on Parkinson’s disease (PD) onset and pathology

MTHFR, methylenetetrahydrofolate reductase; MTRR, methionine synthase reductase.
Table 2. Role of dietary intake of B-vitamins on Parkinson’s disease (PD) onset and progression

l-DOPA, levodopa or l-3,4-dihydroxyphenylalanine.
Table 3. Impact of methyl groups generated by folate metabolism on Parkinson’s disease (PD) patients

COMTi, catechol-O-methyltransferase inhibitor; N/A, not available; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; SNCA, α-synuclein gene; SNc, substantia nigra pars compacta.
Table 4. Impact of levodopa or l-3,4-dihydroxyphenylalanine (l-DOPA) on B-vitamin metabolism in Parkinson’s disease (PD)-affected patients

N/A, not available; MTHFR, methylenetetrahydrofolate reductase; COMTi, catechol-O-methyltransferase inhibitor; SAM, S-adenosylmethionine.
Table 5. Mechanisms through which folate metabolism may be involved in Parkinson’s disease onset and progression

FADD, folic acid-deficient diet; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; SNc, substantia nigra pars compacta; DA, dopaminergic; i.p., intraperitoneal; l-DOPA, levodopa or l-3,4-dihydroxyphenylalanine; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; COMT, catechol-O-methyltransferase.
Results
Polymorphisms in one-carbon metabolism may increase risk for Parkinson’s disease development
Genetic factors may contribute to homocysteine accumulation through polymorphisms in enzymes involved in one-carbon metabolism. The data from our literature search are summarised in Table 1. One of the most common of these is a polymorphism in MTHFR 677C→T, which has been observed at greater frequencies in PD patients than the general population in some studies(Reference Gorgone, Currò and Ferlazzo24,Reference de Lau, Koudstaal and Van Meurs42) , while no differences have been seen in others(Reference Białecka, Kurzawski and Roszmann43). An Italian case–control study found that the 677TT polymorphism was significantly more frequent in PD patients than controls, after controlling for age, sex, folate and vitamin B12 status(Reference Gorgone, Currò and Ferlazzo24). In a prospective cohort study in the Netherlands, the TT variant was associated with increased risk for PD in individuals who smoke(Reference de Lau, Koudstaal and Van Meurs42). A study conducted in Poland found no differences in the distribution of MTHFR alleles in PD compared with control groups(Reference Białecka, Kurzawski and Roszmann43). Regardless of their association with PD, polymorphisms in MTHFR such as 677C→T have been linked to elevated homocysteine concentrations in individuals, including those with PD(Reference de Lau, Koudstaal and Van Meurs42–Reference Zhu, Zhu and He45). It was also demonstrated that the T allele, along with disease status, influenced homocysteine levels in participants, and that significant interaction between these two factors existed(Reference Białecka, Kurzawski and Roszmann43). Results from meta-analysis have also supported a relationship, finding an increased susceptibility associated with the T allele(Reference Zhu, Zhu and He45,Reference Wu, Ding and Sun46) .
Other enzymes involved in one-carbon metabolism have also been shown to be affected, for example, MTRR, which maintains adequate levels of cobalamin during the conversion of homocysteine into methionine(Reference Fong, Shyu and Shieh47). MTRR 1049GG was significantly associated with PD in a Chinese population in a case–control study(Reference Fong, Shyu and Shieh47). These results suggest that there is an increasing risk for PD with the number of polymorphisms that interfere with normal one-carbon metabolism. It therefore appears that such polymorphisms in enzymes involved in one-carbon metabolism may contribute in some way to PD development.
Role of B-vitamins in Parkinson’s disease onset and progression
Several studies have investigated the impact of dietary B-vitamins on PD development; the results are summarised in Table 2. A study in Mexico examined the nutritional status of patients with PD(Reference López-Botello, Gonzalez-Pena and Berrun-Castonon48). Patients reported both losing weight (73 %) and decreased food intake (67 %), which may have been the result of PD itself or of l-DOPA use. PD patients in Japan also had lower folate intake than controls(Reference Murakami, Miyake and Sasaki49). After adjusting for potential confounding factors, folate, vitamin B12 and riboflavin intakes were not associated with PD risk(Reference Murakami, Miyake and Sasaki49). This is worth noting, as it has been observed in other studies that the increased homocysteine levels of PD patients may be due in significant part to low folate levels(Reference dos Santos, Busanello and Miglioranza33). In one such study, PD patients with folate levels below 13 ng/ml had elevated homocysteine compared with controls, while those with folate concentrations above 13 ng/ml showed no significant difference with respect to controls(Reference dos Santos, Busanello and Miglioranza33). Such lower levels of B-vitamins may contribute to increased homocysteine levels in PD patients, either on their own or through a loss of potential protective effects against homocysteine increases precipitated by other factors.
In advanced PD, patients have lower levels of serum vitamin B12, which can lead to further neuropathy and cognitive impairment(Reference Christine, Auinger and Joslin50,Reference Shen51) . Lower B12 levels during early PD have also been reported to result in more mobility issues in patients, whereas higher levels of homocysteine resulted cognitive decline in patients(Reference Christine, Auinger and Joslin50). Further studies have investigated whether dietary intake of B-vitamins may reduce the risk of PD. One such study in the USA examined intake of dietary folate in addition to vitamins B6 and B12 and their relationship to the risk of PD(Reference Chen, Zhang and Schwarzschild52). They found that higher dietary intake of B-vitamins was not associated with lower risk of PD. A study in the Netherlands also found no effects of dietary folate or vitamin B12 on PD risk(Reference de Lau, Koudstaal and Witteman53). However, an association for vitamin B6 was found, but was significant only in smokers. These findings are also supported by a meta-analysis, which found an overall decreased risk of PD with high vitamin B6 intake (OR 0·65; 95 % CI 0·30, 1·01)(Reference Shen51).
It is important to consider that such studies are often based on diet questionnaires, which may not provide completely accurate data regarding dietary and supplement consumption and can be subject to recall error and bias(Reference Shim, Oh and Kim54–Reference Bates, Prentice and Van Der Pols57). Some countries also have mandated folic acid fortification of grains, which would increase baseline levels for their citizens(Reference Castillo-Lancellotti, Tur and Uauy58,Reference Plumptre, Masih and Ly59) . Additionally, consumption is not necessarily equivalent to circulating levels due to differences in metabolic processes between individuals. As a result, future studies may wish to combine such questionnaires regarding intake with physiological measurements such as plasma samples to provide a more accurate picture of dietary factors. Studies including sufficient numbers of participants are also critical to parse potential interactive effects of l-DOPA on homocysteine levels.
A non-randomised trial examined the impact of folic acid supplementation on patients with PD(Reference Ibrahimagic, Smajlovic and Dostovic60). Periodic supplementation with 5 mg/d folic acid for 1–2 months every 6 months reduced homocysteine levels in all patients studied(Reference Ibrahimagic, Smajlovic and Dostovic60). If patients discontinued use for 2 months, this effect remained intact for 90 % of patients. After 4 months, homocysteine would return to elevated levels. This study had a small number of participants and failed to consider other factors such as MTHFR polymorphism or dietary B-vitamin intake but provides a basis for further investigation into whether B-vitamin supplementation may aid in reducing homocysteine levels, which may then lessen its harmful effects.
Reduced methyl groups generated from folate metabolism and impact on gene expression of Parkinson’s disease-specific genes
Effects on methylation in PD patients are also of interest in the study of the disease. The results of our findings regarding methylation effects are summarised in Table 3. As discussed in previous sections, folates and other B-vitamins are linked to methylation reactions through one-carbon metabolism, particularly through effects on levels of the methyl donor SAM. The potential impact of changes in methylation patterns in PD has been reviewed previously(Reference Miranda-Morales, Meier and Sandoval-Carrillo61). Briefly, DNA methylation can block the recognition of binding sites by transcription factors, impairing gene expression by interfering with transcription. Additionally, other proteins can recognise methylated sites and recruit repressor proteins that then lead to gene repression(Reference Miranda-Morales, Meier and Sandoval-Carrillo61).
A case–control study investigated methylation patterns of the human gene for α-synuclein (SNCA), and found that methylation of SNCA intron 1 led to a decrease in gene expression, while inhibiting methylation activated expression(Reference Jowaed, Schmitt and Kaut62). Methylation potential in patients with PD was reduced at this site in SNc, putamen and cortex samples compared with controls. When an inhibitor of DNA methylation was applied, methylation of the SNCA region in question was specifically reduced, and increased amounts of both α-synuclein mRNA and protein were produced.
The potential clinical consequences of reduced methylation in PD patients were investigated in another case–control study(Reference Obeid, Schadt and Dillmann35). Levels of SAM and α-synuclein were shown to be inversely related in this study, with low levels of SAM associated with increased α-synuclein. Improved cognitive ability was also associated with higher methylation potential, as assessed by higher SAM:SAH ratios. The researchers offered two possible explanations for their findings. They suggest that decreased SAM leads to hypomethylation of SNCA, which results in greater α-synuclein expression. This is consistent with the findings of Jowaed et al.(Reference Jowaed, Schmitt and Kaut62). Second, they propose that increased SAH may act to inhibit processes involved in the repair of damaged proteins such as α-synuclein. Damaged α-synuclein is prone to aggregation, thus this action would also lead to increased accumulation. Therefore, the potential effects of homocysteine in PD extend beyond contributions to neurodegeneration and oxidative stress and may also influence gene expression and pathological protein aggregation.
Treatment with l-DOPA (levodopa (l-3,4-dihydroxyphenylalanine)) makes an impact on one-carbon metabolism
l-DOPA is the most widely used treatment for PD and has been reported to affect folate metabolism(Reference Wurtman37,Reference Moskowitz and Wurtman38) . Data are summarised in Table 4. Numerous studies have demonstrated that patients with PD consistently show elevated levels of homocysteine compared with controls(Reference Rodriguez-Oroz, Lage and Sanchez-Mut31–Reference Obeid, Schadt and Dillmann35,Reference Kuhn, Roebroek and Blom40,Reference Müller, Werne and Fowler41,Reference Allain, LeBouil and Coredilet63) . However, it remains uncertain if this is due to PD, as additional research has indicated that l-DOPA can also contribute to elevated homocysteine. l-DOPA increases the amount of homocysteine in circulation by receiving a methyl group provided by SAM, converting SAM to SAH which is then readily hydrolysed into homocysteine(Reference Brosnan, Jacobs and Stead64,Reference Liu, Wilson and Charlton65) . This reaction involves the enzyme catechol-O-methyltransferase (COMT). Because l-DOPA is the most widely used treatment for PD, this no doubt has an impact on measurements taken from PD patient populations, as the majority are taking this drug. Indeed, several studies have found that homocysteine levels were significantly higher in PD patients treated with l-DOPA compared with non-treated individuals(Reference Yuan, Sheu and Yu25,Reference De Bonis, Tessitore and Pellecchia44,Reference Miller, Selhub and Nadeau66) . A case–control study examined PD patients before receiving l-DOPA compared with controls, and observed no significant difference in homocysteine levels(Reference Yasui, Nakaso and Kowa67). After patients began receiving l-DOPA, all but one individual experienced a significant increase in plasma homocysteine. Researchers also observed that the increases were most notable for those with the 677C→T MTHFR polymorphism. Relative increases of 35·0 (sd 21·3) % for the CC genotype, 44·8 (sd 36·7) % for CT, and 156·2 (sd 108·5) % for the TT genotype were observed(Reference Yasui, Nakaso and Kowa67). The high standard deviations are probably due to the small numbers of participants in this study, which had only twenty participants in total, and six, eight and six patients with the CC, CT and TT genotypes, respectively. Nevertheless, this study indicates a potential influence of polymorphisms in folate metabolism on the effects of l-DOPA on homocysteine levels. Another study investigating the impact of l-DOPA on PD patients found that homocysteine levels were elevated in TT genotype participants and that homocysteine levels were negatively correlated with serum folate levels(Reference Yasui, Kowa and Nakaso68).
Such an impact as a result of additional factors has also been suggested in other studies. For example, another case–control study reported that elevated levels of homocysteine in PD patients compared with controls were affected by multiple factors, including PD, MTHFR 677C→T polymorphism, B-vitamin intake and folate level(Reference Camicioli, Bouchard and Somerville32). When total homocysteine levels were examined in cerebrospinal fluid of PD patients both before and after l-DOPA treatment and in healthy controls, it was reported that PD patients had significantly higher levels of homocysteine than controls before treatment, and levels after treatment were significantly higher than before treatment in those with PD(Reference Isobe, Abe and Terayama69). The difference in the total homocysteine concentration before and after treatment was also greater for those who took higher doses of l-DOPA (450 v. 300 mg/d), again indicating that l-DOPA is affecting homocysteine levels. Conversely, another study examined patients on different dosages of l-DOPA in comparison with controls, including non-treated patients, and found that while PD patients had elevated levels of homocysteine, levels did not depend on the dosage of l-DOPA that patients were receiving(Reference Religa, Czyzewski and Styczynska70). Also observed was a trend toward higher homocysteine levels in non-treated PD patients compared with controls(Reference Religa, Czyzewski and Styczynska70), though there were only a small number of participants in this group (fifteen compared with ninety-nine l-DOPA-treated patients). They also found that disease duration significantly influenced homocysteine levels(Reference Religa, Czyzewski and Styczynska70). These results tend to indicate the potential for interaction between l-DOPA use, polymorphisms in folate metabolism, B-vitamin intake, and disease duration in the development and progression of PD. Based on these findings, PD patients who have been administered l-DOPA for a long period of time may benefit from supplementation of folic acid or other B-vitamins(Reference Müller, Werne and Fowler41,Reference Miller, Selhub and Nadeau66) .
Another approach that researchers have employed to disentangle the potential effects that l-DOPA has on homocysteine is examining the effects of COMT inhibitors in conjunction with l-DOPA treatment. As mentioned earlier, COMT is involved in the O-methylation of l-DOPA that converts SAM into SAH, which is further hydrolysed into homocysteine (Fig. 1). If elevated homocysteine levels are due to increased amounts produced from SAH through this mechanism, inhibiting COMT should result in a reduction in homocysteine levels compared with individuals taking only l-DOPA. Results from such investigations have been conflicting. In one study, the COMT inhibitor, Entacapone, was administered to one group of PD patients receiving l-DOPA, while another received l-DOPA alone(Reference Kocer, Guven and Comoglu71). No significant differences in homocysteine levels were observed between the groups. Similar results were reported in another study that found that COMT inhibitors did not significantly rescue SAM or reduce total homocysteine(Reference Obeid, Schadt and Dillmann35). Such findings suggest that if homocysteine levels are in fact elevated by l-DOPA use, this may occur through means other than via COMT-mediated methylation of l-DOPA. However, several key significant differences were observed between the treatment groups, including age, disease duration and age at disease onset, which may have had confounding effects on homocysteine levels.
In another study, three groups consisting of PD patients treated with l-DOPA, PD patients treated with l-DOPA and a COMT inhibitor, and controls were examined(Reference Lamberti, Zoccolella and Iliceto72). Plasma levels of homocysteine were significantly increased in both groups of PD patients compared with controls. When compared with each other, those treated with a COMT inhibitor in addition to l-DOPA had significantly lower levels of homocysteine compared with those who received l-DOPA only. There were also fewer patients in this group that had pathologically high (over 20 μmol/l) levels of homocysteine, with only 5 % compared with 30 % of those in the l-DOPA only group (the control group had none). An important consideration and potential confounding factor in this study was that folate concentrations were found to be significantly lower in the group treated with l-DOPA alone. As discussed previously, low levels of folate are associated with high homocysteine levels. Additionally, other studies have found no impact of l-DOPA on homocysteine levels, suggesting that l-DOPA may be interacting with other factors, such as folate deficiency or the duration of the disease, to elicit effects on homocysteine(Reference dos Santos, Busanello and Miglioranza33). This theory is supported by further research that has proposed that the extent of elevated homocysteine in PD patients can be influenced by their folate status(Reference Miller, Selhub and Nadeau66). However, the researchers performed additional analysis that claimed that the homocysteine elevations observed in l-DOPA-alone group were due to the absence of a COMT inhibitor, rather than decreased folate levels(Reference Lamberti, Zoccolella and Iliceto72). Nevertheless, it will prove important to design future studies to control for differences in B-vitamin status, along with other potential confounding factors, in order to determine the precise roles of each on PD development and progression. Overall, these findings suggest that while treatment with l-DOPA probably has an impact on homocysteine levels, it cannot be ruled out entirely that factors such as PD contribute to observed elevations. l-DOPA also interacts with the trans-sulfuration component of one-carbon metabolism. Specifically, with the active form of vitamin B6, pyridoxal-5’-phosphate (P5P), it sequesters P5P from P5P-dependent enzymes including CBS, potentially resulting in a functional B6 deficiency(Reference Evered73,Reference van der Steen, den Heijer and Groen74) .
Mechanisms through which folate metabolism may influence Parkinson’s disease onset and progression
To better understand how one-carbon metabolism may impact PD onset and progression, researchers have turned to in vitro and in vivo model systems. Summaries of the study details are listed in Table 5. In vitro, folate deficiency in cell culture has been shown to elicit neurodegeneration and increase reactive oxygen species (ROS) production(Reference Ho, Ashline and Dhitavat75). Direct exposure of homocysteine to cells also produced the same effects, while inhibition of homocysteine formation was effective at suppressing increases in ROS. Such results reinforce the inverse relationship between folate and homocysteine. In another study, homocysteine exposure in combination with the pesticide rotenone or Fe led to increased membrane depolarisation in the mitochondria of human DA cells(Reference Duan, Ladenheim and Cutler76). Mitochondrial ROS levels also increased with rotenone or Fe exposure and were further exacerbated by homocysteine but were suppressed by treatment with an antioxidant or an inhibitor of DNA damage.
In rodent studies, when folate-deficient diets have been combined with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) exposure, they result in reduced numbers of surviving DA neurons and increased motor impairment compared with a control diet(Reference Duan, Ladenheim and Cutler76). Direct infusion of homocysteine into the SNc has also been shown to produce these same effects(Reference Duan, Ladenheim and Cutler76). Because these observations were seen only when MPTP was administered, and not when homocysteine was administered on its own, this may imply that folate deficiency and elevations in homocysteine act to increase vulnerability to neurodegeneration, rather than triggering cell death directly. However, other experiments have found that homocysteine on its own can prove toxic for DA neurons(Reference Lee, Chen and Soliman77,Reference Imamura, Takeshima and Nakaso78) . In a study investigating the effects on rat primary mesencephalic cells, homocysteine enhanced cell death in response to MPTP in a dose-dependent manner(Reference Imamura, Takeshima and Nakaso78). Cells with intracellular dopamine were also more vulnerable to homocysteine’s toxic effects than other cells. Similarly, Lee et al.(Reference Lee, Chen and Soliman77) found that homocysteine decreased tyrosine hydroxylase immunoreactivity as well as locomotor activity in mice after it was administered chronically over a period of 36 d. These inconsistent findings underline the need for further investigation regarding this topic.
The effects of l-DOPA treatment in mouse models of PD have also provided some insight into how it may affect homocysteine accumulation. Acute v. chronic treatment has been reported to increase homocysteine and reduce levels of SAM(Reference Taufek and Bone79–Reference Wagner, Danzin and Huot-Olivier81). Furthermore, when animals were placed on a folic acid-deficient diet there was a more robust response to l-DOPA compared with control animals(Reference Daly, Miller and Nadeau80), suggesting an interaction between folate and l-DOPA metabolism. In non-MPTP- and MPTP-treated mice, chronic treatment with l-DOPA leads to significant increases in homocysteine in the SNc(Reference Bhattacharjee, Khairujjaman Mazumder and Paul82). l-DOPA also significantly increased the level of homocysteine in the SNc in MPTP-treated mice compared with the non-MPTP mice that also received l-DOPA. Researchers also examined the effect that these treatments had on DA cells in the SNc. They observed a 51 % reduction in TH+ cells for the MPTP plus l-DOPA group v. 47 % for MPTP alone. Therefore, the significant increase in homocysteine in the former did not result in a significantly greater loss of DA cells in the SNc. This is in contrast to the results seen by Duan et al.(Reference Duan, Ladenheim and Cutler76), who observed an increase in neurodegeneration following MPTP when homocysteine was increased using folate-deficient diets or was directly infused(Reference Duan, Ladenheim and Cutler76). These differences may be due to differences in MPTP dosages between the studies, or the length of time between the last MPTP treatment and euthanisation(Reference Duan, Ladenheim and Cutler76,Reference Bhattacharjee, Khairujjaman Mazumder and Paul82) . Of course, l-DOPA may also have had additional effects in the latter study that affected the degeneration of DA cells. However, another study also examined mice after more than 30 d of homocysteine injections, and observed significantly increased homocysteine levels in the striatum, impaired locomotor activity, and reduced tyrosine hydroxylase immunoreactivity in the SNc(Reference Lee, Chen and Soliman77). Mouse models have also been used to study l-DOPA’s impact on methylation processes. Administration of l-DOPA increased the amount of SAH, decreased SAM, and decreased SAM:SAH ratios in one study(Reference Liu, Wilson and Charlton65). The brain is capable of de novo synthesis of methyl groups; however, when animals are deficient in dietary folic acid combined with l-DOPA, this results in a significant decrease in methionine levels in brain tissue and serum(Reference Ordonez and Wurtman83). Another study demonstrated that blocking COMT with the inhibitor Ro 41-0960 can prevent the l-DOPA changes, such as decreased SAM and increased SAH and homocysteine levels in a rat model(Reference Miller, Shukitt-Hale and Villalobos-Molina84). Furthermore, methionine intake can reverse the impact of l-DOPA in methyl group depletion(Reference Taufek and Bone79). This lends support to work demonstrating the potential for l-DOPA to influence gene expression.
Despite the lack of clear evidence suggesting that B-vitamin intake reduces risk of PD, the effects of B-vitamin supplementation on PD pathology have also been investigated. In one study, the effect of supplementation using several B-vitamin doses in several combinations was examined both in terms of behaviour and effect on levels of homocysteine in a rat model of PD(Reference Haghdoost-Yazdi, Fraidouni and Faraji85). 6-Hydroxydopamine, a well-characterised model of DA degeneration, was administered to the striatum. Rats that received ten times the folate normally given in a minimum essential medium diet had improved performance on rotational behaviour testing and rotarod(Reference Haghdoost-Yazdi, Fraidouni and Faraji85). The groups receiving five times the combination of B-vitamins also performed at levels close to control on the rotarod. Interestingly, levels of homocysteine were not reduced in any of the groups, and were in fact elevated in comparison with controls(Reference Haghdoost-Yazdi, Fraidouni and Faraji85). This unexpected finding indicates that such supplementation is providing beneficial effects through means other than decreasing homocysteine concentrations. Another study examined the effect of supplemental folate in a Drosophila model of early-onset familial PD(Reference Srivastav, Singh and Yadav86). Researchers introduced a novel recessive allele for the parkin gene, which resulted in reduced mRNA and null amounts of the Parkin protein. Parkin has a role in the degradation of unfolded proteins and is linked to normal mitochondrial function. The absence of Parkin resulted in numerous deficiencies in homozygous flies, including increased pupal lethality, decreased progression to later life stages and impaired motor function(Reference Srivastav, Singh and Yadav86). It also increased oxidative stress, while reducing antioxidant activity and mitochondrial functionality, as evidenced by lower levels of ATP(Reference Srivastav, Singh and Yadav86). Individuals given a 10–250 µm effective dose of folic acid showed at least partial recovery. Reductions in lethality, greater numbers of flies transitioning to later life stages, and improved motor function were observed. Oxidative stress decreased and the amount of ATP present increased, indicating improved mitochondrial function. These observations suggest that folate supplementation may be useful for attenuating some of the effects of DA degeneration in PD.
Discussion
The present review assessed the role of one-carbon metabolism and homocysteine on the onset and progression of PD, as well as potential mechanisms of action. Our findings suggest that genetic polymorphisms in one-carbon metabolism may play a role in the risk of developing the disease, possibly through increased levels of homocysteine. Dietary levels of B-vitamins such as folate may be reduced in PD-affected patients. However, any benefit of B-vitamin supplementation for PD patients in general remains undetermined. To replenish the loss of dopamine, l-DOPA is frequently administered to PD-affected patients, leading to increased levels of homocysteine. For this reason, supplementation with B-vitamins may be beneficial for PD patients taking l-DOPA. Folate metabolism is also involved in generating methyl groups that are used for methylation reactions and gene expression. The literature suggests that there are methylation changes in PD-specific genes which may affect PD progression. The mechanisms through which folate metabolism makes an impact on PD progression remain undefined, but a consensus in the literature suggests that deficiencies in folate can increase vulnerability to damage.
Recent epidemiological data have suggested that homocysteine may be involved in the onset and progression of neurodegenerative diseases(Reference Hannibal and Blom87). Using in vitro models, the link between homocysteine and neurodegenerative disease may be indicated by increased DNA damage(Reference Kruman, Culmsee and Chan88,Reference Kruman, Kumaravel and Lohani89) , oxidative stress(Reference Vanzin, Biancini and Sitta90–Reference Kim and Pae93) and apoptosis(Reference Baydas, Reiter and Akbulut94). The concentration of homocysteine used in many in vitro studies ranges from 0·5 to 300 µm(Reference Kruman, Culmsee and Chan88,Reference Lipton, Kim and Choi91,Reference Kim and Pae93) . It is important to note that the levels of homocysteine used in in vitro studies do not match the physiological levels (about 4 to 10 µm) present in living systems(Reference Rodriguez-Oroz, Lage and Sanchez-Mut31,Reference Camicioli, Bouchard and Somerville32) . The association between high levels of homocysteine and PD is unclear(Reference Rodriguez-Oroz, Lage and Sanchez-Mut31,Reference Camicioli, Bouchard and Somerville32,Reference Kocer, Guven and Conkbayir95) . Supplementation of B-vitamins has been reported to result in amelioration of symptoms associated with PD in both human(Reference Ibrahimagic, Smajlovic and Dostovic60) and model system studies(Reference Srivastav, Singh and Yadav86). In animal studies, there is a minimal reduction in homocysteine levels(Reference Haghdoost-Yazdi, Fraidouni and Faraji85). These reports suggest that homocysteine may be a marker for disease state and not the cause of disease onset and progression. Increased levels of homocysteine can be a result of deficiencies in folate metabolism, the nutrient choline, as well as reduced activity in the trans-sulfuration pathway.
PD is a complex disease and the cause of the loss of DA cells is not well understood. Both genetics(Reference Dawson, Ko and Dawson96) and dietary factors(Reference Seidl, Santiago and Bilyk17) have been implicated in disease pathology. Folates play an important role in the brain because of their functions in nucleotide synthesis, DNA repair, methylation, membrane lipid metabolism, second messenger signalling, ion channels and neurotransmitter and protein synthesis(Reference Murray, Emmerson, Jadavji and Lee20). The elderly population is often deficient in B-vitamins, such as folate, riboflavin, vitamin B12 and vitamin B6(Reference Fenech97,Reference Jannusch, Jockwitz and Bidmon98) . This is because of age-related changes in absorption of nutrients from food(Reference Moore, Hughes and Ward99). Supplementation with B-vitamins may be necessary for PD-affected individuals to sustain optimal levels of these vitamins. B-vitamins are also involved in the generation of methyl groups and SAM; therefore changes in gene expression may be affecting disease progression. Because of the many roles that B-vitamins have within the brain, low intake or status could render cells in the brain, specifically DA cells, vulnerable to degeneration. For PD patients with high levels of homocysteine, B-vitamin supplementation should be considered. This point is particularly important for PD patients taking l-DOPA, which can further increase homocysteine levels.
Conclusion and future directions
Our analysis of clinical and basic science studies suggests that the data still remain unclear; one-carbon metabolism and homocysteine may be involved in PD onset, pathology and progression, but the precise relationships have yet to be determined. Future research using more clinically relevant model systems that investigate mechanisms of action using environmental toxin PD models, as well as randomised controlled studies in human subjects to understand whether dietary supplementation is beneficial for PD patients, will provide more insight.
Author ORCIDs
Nafisa Jadavji 0000-0002-3557-7307
Acknowledgements
The authors would like to thank Marc T. Avery for his useful comments on the manuscript.
This research was supported by a Canadian Institutes of Health Research studentship (L. K. M.) and a Natural Sciences and Engineering Research Council of Canada fellowship (N. M. J.).
Each author read and approved the final submitted manuscript. L. K. M. conducted the introduction, human bibliographic research and wrote the manuscript. N. M. J. conceptualised the study, conducted the model system bibliographic research, interpreted the data, supervised, reviewed and wrote the final draft of the manuscript.
There are no conflicts of interest.






