


INTRODUCTION
The phylum of Apicomplexa contains approximately 6000 unicellular, eukaryotic parasites including Plasmodium spp., the causative agent of Malaria, and Toxoplasma gondii, responsible for toxoplasmosis in many important farm animals and humans (Sato, Reference Sato2011). Morphologically, all members of the apicomplexan family share a distinctive apical complex, together with species dependent apical-localized organelles (McFadden and Yeh, Reference McFadden and Yeh2017). These parasites employ complex life cycles including both sexual and asexual reproduction. Furthermore, in many cases their life cycles involve multiple hosts. T. gondii, first described in 1908 and often regarded as one of the most successful apicomplexan parasites, represents the key model organism of the phylum (Dubey, Reference Dubey2008; Weiss and Dubey, Reference Weiss and Dubey2009; Szabo and Finney, Reference Szabo and Finney2017). Its primary hosts are members of the Felidae (cats) family while all other warm-blooded animals, including humans, are intermediate hosts. It is estimated that up to one third of the human population is infected with T. gondii and thus are potential carriers. Although the infection is usually asymptotic in healthy individuals it can cause severe congenital disease during pregnancy (Kaye, Reference Kaye2011), and lead to life-threatening infections in immuno-compromised patients including those suffering from HIV, receiving an organ transplant or undergoing cancer chemotherapy treatment (Flegr et al. Reference Flegr, Prandota, Sovickova and Israili2014). Current toxoplasmosis treatment options are limited to a handful of antimicrobials such as sulphonamides, folic acid derivatives and certain macrolide antibiotics. However, these drugs often show limited efficacy and are associated with significant side effects (Alday and Doggett, Reference Alday and Doggett2017). Furthermore, there are no treatments available to target tissue cysts, the persistent form in which the parasite evades the host immune system, and to eradicate persistent T. gondii infections (Opsteegh et al. Reference Opsteegh, Kortbeek, Havelaar and van der Giessen2015). Therefore, new drug targets and novel therapies are urgently needed. In addition to high-throughput screening approaches (Norcliffe et al. Reference Norcliffe, Alvarez-Ruiz, Martin-Plaza, Steel and Denny2014), structure-based methods in close combination with medicinal chemistry and biophysical and biological validation have become powerful tools in the search of new drugs and treatments (Hunter, Reference Hunter2009; Verlinde et al. Reference Verlinde, Fan, Shibata, Zhang, Sun, Deng, Ross, Kim, Xiao, Arakaki, Bosch, Caruthers, Larson, Letrong, Napuli, Kelly, Mueller, Zucker, Van Voorhis, Buckner, Merritt and Hol2009; Groftehauge et al. Reference Groftehauge, Hajizadeh, Swann and Pohl2015; Hol, Reference Hol2015; Muller, Reference Muller2017).
THE ROLE OF CALCIUM-DEPENDENT PROTEIN KINASES (CDPKs)
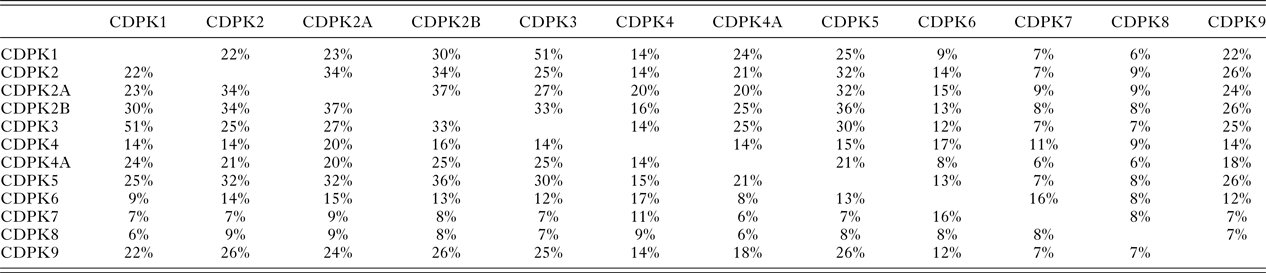
In T. gondii Ca2+-ions play key roles in cell signalling and in pathogen–host interactions including cell invasion, motility of the parasite within the host and differentiation during the parasites complex life cycle (Irvine, Reference Irvine1986; Nagamune et al. Reference Nagamune, Moreno, Chini and Sibley2008; Lourido and Moreno, Reference Lourido and Moreno2015). CDPKs are a family of serine/threonine kinases that are only found in plants and protists including ciliates and apicomplexan parasites. Importantly, these kinases provide the mechanistic link between calcium signalling and motility, differentiation and invasion (Tzen et al. Reference Tzen, Benarous, Dupouy-Camet and Roisin2007; Billker et al. Reference Billker, Lourido and Sibley2009). These crucial roles of CDPKs have been proven through a range of knock-out studies in various species and underline the potential of CDPKs as targets for novel therapeutics (Long et al. Reference Long, Wang and Sibley2016). CDPKs are members of the Calmodulin/Calcium kinase (CaM) family. They all share an N-terminal kinase domain (KD) linked via a junctional domain to a series of C-terminal Calcium-binding motifs. In T. gondii at least 12 different CDPKs have been putatively identified ranging in size from 507 (CDPK1) to more than 2000 amino acids (CDPK7, CDPK80) (Morlon-Guyot et al. Reference Morlon-Guyot, Berry, Chen, Gubbels, Lebrun and Daher2014). Although there is probably overlap in functionalities, different sub-cellular locations and varying expression levels during the parasites’ life cycle is likely to lead to different biological functions within the CDPK family (Hui et al. Reference Hui, El Bakkouri and Sibley2015). The shared sequence identities range from 51% (CDPK1 and CDPK3) (Treeck et al. Reference Treeck, Sanders, Gaji, LaFavers, Child, Arrizabalaga, Elias and Boothroyd2014) to lower than 10% (Table 1). These variations in length and sequence support the notion that members of the CDPK family act upon a range of substrates and fulfil different functions in T. gondii biology. Recent knock-out studies using CRISPR-Cas9 indicate that CDPK4, CDPK5, CDPK6, CDPK8 and CDPK9, respectively, have no effect on virulence and on normal growth (Wang et al. Reference Wang, Huang, Li, Chen, Ning and Zhu2016), however, knock-down studies have shown that CDPK7 is crucial for survival due to a critical role in parasite division (Morlon-Guyot et al. Reference Morlon-Guyot, Berry, Chen, Gubbels, Lebrun and Daher2014). More detailed studies have been performed on the smaller family members. CDPK3 with 537 amino acids has been implicated in motility and host cell egress (McCoy et al. Reference McCoy, Stewart, Uboldi, Li, Schroder, Scott, Papenfuss, Lehane, Foster and Tonkin2017). CDPK2 (711 amino acids) has been shown to act as key regulator of amylopectin metabolism (Uboldi et al. Reference Uboldi, McCoy, Blume, Gerlic, Ferguson, Dagley, Beahan, Stapleton, Gooley, Bacic, Masters, Webb, McConville and Tonkin2015). The loss of CDPK2 results in the build-up of amylum with fatal consequences for T. gondii in its chronic stage. Importantly, this family member contains an N-terminal carbohydrate-binding domain that may offer new opportunities for drug design (Uboldi et al. Reference Uboldi, McCoy, Blume, Gerlic, Ferguson, Dagley, Beahan, Stapleton, Gooley, Bacic, Masters, Webb, McConville and Tonkin2015). CDPK1, which is mainly located in the cytosol, has been shown to be required for the microneme secretion at the apical complex and parasite proliferation. The molecular mechanism, however, remains elusive (Lourido et al. Reference Lourido, Shuman, Zhang, Shokat, Hui and Sibley2010; Child et al. Reference Child, Garland, Foe, Madzelan, Treeck, van der Linden, Oresic Bender, Weerapana, Wilson, Boothroyd, Reese and Bogyo2017). Here we will review strategies and recent results in the discovery, design and potency of inhibitors targeting the KD of CDPK1 from T. gondii (TgCDPK1).
Table 1. The protein sequence identities between the 12 putative calcium-dependent protein kinases (CDPKs) of T. gondii, rounded to the nearest whole number, derived from a multiple sequence alignment (MSA) generated using Clustal Omega (Sievers et al. Reference Sievers, Wilm, Dineen, Gibson, Karplus, Li, Lopez, McWilliam, Remmert, Söding, Thompson and Higgins2011)
ACTIVATION OF TgCDPK1 BY CALCIUM
The mechanism of activation and inhibition was unravelled in 2010 when the crystal structures of both the auto-inhibited and the Ca2+-activated forms of TgCDPK1 were published (Ojo et al. Reference Ojo, Larson, Keyloun, Castaneda, Derocher, Inampudi, Kim, Arakaki, Murphy, Zhang, Napuli, Maly, Verlinde, Buckner, Parsons, Hol, Merritt and Van Voorhis2010; Wernimont et al. Reference Wernimont, Artz, Finerty, Lin, Amani, Allali-Hassani, Senisterra, Vedadi, Tempel, Mackenzie, Chau, Lourido, Sibley and Hui2010). These structures revealed the expected KD in similar overall conformations, however, the Ca2+-binding domain (also designated CPDK activating domain or CAD) adopted two vastly different conformations and orientations (Fig. 1). In its inactive state the CAD (shown in rasberry red) adopts an elongated form reminiscent of apo-calmodulin starting with a long helix followed by the first Ca2+-binding motifs (EF-hands), which is connected via another long helix to the second pair of C-terminal EF-hands (Fig. 1a). The first long helix has been suggested to be responsible for the auto-inhibitory effect by blocking the substrate binding site and providing a basic lysine residue to bind a cluster of conserved acidic residues. However, this may not be the only mechanism of deactivation as it has more recently been shown that removal of the regulatory domain alone does not lead to an active KD (Ingram et al. Reference Ingram, Knockenhauer, Markus, Mandelbaum, Ramek, Shan, Shaw, Schwartz, Ploegh and Lourido2015). The CAD domain activated by Ca2+-binding appears to be required to maintain the KD in its active conformation. Calcium binding leads to a dramatic rearrangement and refolding of the protein chain (Fig. 1b) (Wernimont et al. Reference Wernimont, Artz, Finerty, Lin, Amani, Allali-Hassani, Senisterra, Vedadi, Tempel, Mackenzie, Chau, Lourido, Sibley and Hui2010). The entire regulatory domain is shifted to the other side of the protein hence liberating the active site of the KD as shown in Fig. 1c. In addition, the regulatory calcium-binding domain is collapsed so that the two long helices are no longer arranged in an anti-parallel fashion but are partially unwound and interwoven to form a more globular overall shape. These structural changes are reminiscent to the calcium-bound structure of calmodulin (Kursula, Reference Kursula2014).

Fig. 1. Ribbon representation of the crystal structure of CDPK1 from T. gondii with the kinase domain depicted in cyan, the regulatory domain in raspberry red. (a) CDPK1 in its inactive auto-inhibited state (PDB code: 3KU2) (Wernimont et al. Reference Wernimont, Artz, Finerty, Lin, Amani, Allali-Hassani, Senisterra, Vedadi, Tempel, Mackenzie, Chau, Lourido, Sibley and Hui2010). (b) CDPK1 in its calcium-bound, activated state with the Ca2+-ions shown as green spheres and the non-hydrolysable ligand ANP in stick representation (PDB code: 3HX4) (Wernimont et al. Reference Wernimont, Artz, Finerty, Lin, Amani, Allali-Hassani, Senisterra, Vedadi, Tempel, Mackenzie, Chau, Lourido, Sibley and Hui2010). (c) Ribbon diagram of the least-squares superposition of the inactive and active forms of TgCDPK1 with the kinase domains shown in cyan (active) and blue (inactive), the regulatory domain in shades of red, respectively. Only the kinase domain was used to calculate the transformation matrix, which was then applied to the entire protein chain. CDPKs, calcium-dependent protein kinases.
COMPARISON WITH HUMAN KINASES
Historically, characterising (protozoan) kinases as potential drug targets and developing selective inhibitors has been considered challenging due to the fact that the overall protein fold and the active sites of all kinases are structurally well conserved (Scapin, Reference Scapin2002). The structural similarities of the KD are obvious when comparing the crystal structures of the KD of TgCDPK1 with the Calcium/Calmodulin (CaM) dependent-kinase II from H. sapiens (HsCaMKII) (Fig. 2a) (Rellos et al. Reference Rellos, Pike, Niesen, Salah, Lee, von Delft and Knapp2010). These two proteins, which share a sequence identity of approximately 42% over 264 residues of the KD, display the same canonical kinase fold and superimpose with an overall root mean square deviation (rmsd) of approximately 1·5 Å. Note that the loop over the adenosine triphosphate (ATP) binding site adopts a very different conformation presumably due to an induced fit of binding of two very different ligands. TgCDPK1 is bound to the ATP analogue ANP (Fig. 2a) while HsCaMKII is bound to a comparatively small inhibitor. More importantly there are significant differences in the ATP binding site, specifically a residue with no side chain (glycine) close to the adenine binding position. This residue, Gly128 is also termed the gatekeeper residue. Almost all mammalian kinases possess a large residue, a phenylalanine in HsCaMKII for example, in this position. Hence, CDPK1 feature an enlarged ATP binding site with a hydrophobic pocket that can be exploited for structure-based drug design. This key structural difference in the binding pocket is shown in the surface representation where the ATP-analogue is shown as stick representation (Fig. 2b). The additional space at the end of the pocket below the surface of the gatekeeper residue Gly 128 in magenta is clearly visible.

Fig. 2. (a) Least squares superposition of the kinase domain of TgCDPK1 (depicted in cyan) in its active form with HsCaMKII bound to an inhibitor (PDB: 2VZ6) (shown in orange) (Rellos et al. Reference Rellos, Pike, Niesen, Salah, Lee, von Delft and Knapp2010). The non-hydrolysable ATP analogue bound in TgCDPK1 is presented as ball-and-stick representation to highlight the substrate binding site. (b) Surface representation of TgCDPK1 viewing into the binding pocket with colour coding according to atom type (oxygen in red, nitrogen in blue, carbon in grey). The surface of Gly128 (gatekeeper residue) is shown in magenta at the top of the figure highlighting the additional space in the binding pocket. ATP, adenosine triphosphate; CDPKs, calcium-dependent protein kinases.
DEVELOPMENT OF SPECIFIC TgCDPK1 INHIBITORS
Soon after the structural differences between TgCDPK1 and the mammalian homologues were identified, two groups started to develop selective TgCDPK1 inhibitors (Ojo et al. Reference Ojo, Larson, Keyloun, Castaneda, Derocher, Inampudi, Kim, Arakaki, Murphy, Zhang, Napuli, Maly, Verlinde, Buckner, Parsons, Hol, Merritt and Van Voorhis2010; Wernimont et al. Reference Wernimont, Artz, Finerty, Lin, Amani, Allali-Hassani, Senisterra, Vedadi, Tempel, Mackenzie, Chau, Lourido, Sibley and Hui2010). Initial compounds were based on known inhibitors previously developed for yeast kinases featuring amino acids with small side chains at the gatekeeper position. Importantly, these known kinase inhibitors, termed bumped kinase inhibitors (BKI) have been shown to be inactive against mammalian kinases (Hanke et al. Reference Hanke, Gardner, Dow, Changelian, Brissette, Weringer, Pollok and Connelly1996). Generally, BKIs are based on the planar pyrazolo[3,4-d]pyrimidin-4-amine substituted with a bulky hydrophobic group on the C3 position (Bishop et al. Reference Bishop, Shah, Liu, Witucki, Kung and Shokat1998). The first example of a BKI with a sub-μmolar IC50 is 1-(1-methylethyl)-3-(naphthalen-1-ylmethyl)-1H-pyrazolo[3,4-d] pyrimidin-4-amine. The co-crystal structure of TgCDPK1 shows that the naphtalen-1-ylmethyl- moiety fills the hydrophobic pocket created by the small gatekeeper residue Gly128 and lined by methionine and leucine residues, and one lysine residue (Fig. 3a and b). The chemically closely related 1-tert-butyl-3-naphthalen-2-yl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (Fig. 3c and d) adopts a similar conformation with the bulky aromatic substituent at the C3 position occupying the space next to the gatekeeper residue. Critically for the subsequent drug development was the fact that these and related BKIs reduced T. gondii proliferation significantly (Ojo et al. Reference Ojo, Larson, Keyloun, Castaneda, Derocher, Inampudi, Kim, Arakaki, Murphy, Zhang, Napuli, Maly, Verlinde, Buckner, Parsons, Hol, Merritt and Van Voorhis2010; Sugi et al. Reference Sugi, Kato, Kobayashi, Watanabe, Kurokawa, Gong, Pandey, Takemae and Akashi2010). These results sparked extensive medicinal chemistry efforts where a large number of compounds based on the BKI scaffold (4-amino-1H-pyrazole[3,4-d]pyrimidine) were synthesized and tested. A number of compounds exhibited sub- or low-nanomolar IC50 values and high activity in parasite growth models (EC50 in the low- and sub-μmolar range) while retaining specificity when compared with mammalian kinases (Lourido et al. Reference Lourido, Jeschke, Turk and Sibley2013; Zhang et al. Reference Zhang, Ojo, Vidadala, Huang, Geiger, Scheele, Choi, Reid, Keyloun, Rivas, Siddaramaiah, Comess, Robinson, Merta, Kifle, Hol, Parsons, Merritt, Maly, Verlinde, Van Voorhis and Fan2014; Moine et al. Reference Moine, Dimier-Poisson, Enguehard-Gueiffier, Logé, Pénichon, Moiré, Delehouzé, Foll-Josselin, Ruchaud, Bach, Gueiffier, Debierre-Grockiego and Denevault-Sabourin2015). In addition to the pyrazolopyrimidine (PP) scaffolds, acylbenzimidazole and 5-aminopyrzazole-4-carboxamide-based compounds have been shown to have similar properties (Fig. 4) (Zhang et al. Reference Zhang, Ojo, Johnson, Larson, He, Geiger, Castellanos-Gonzalez, White, Parsons, Merritt, Maly, Verlinde, Van Voorhis and Fan2012; Zhang et al. Reference Zhang, Ojo, Vidadala, Huang, Geiger, Scheele, Choi, Reid, Keyloun, Rivas, Siddaramaiah, Comess, Robinson, Merta, Kifle, Hol, Parsons, Merritt, Maly, Verlinde, Van Voorhis and Fan2014; Huang et al. Reference Huang, Ojo, Zhang, Rivas, Vidadala, Scheele, DeRocher, Choi, Hulverson, Barrett, Bruzual, Siddaramaiah, Kerchner, Kurnick, Freiberg, Kempf, Hol, Merritt, Neckermann, de Hostos, Isoherranen, Maly, Parsons, Doggett, Van Voorhis and Fan2015). While the initial BKIs showed excellent potency in vitro and in vivo they also exhibited significant hERG (human Ether-Related Gene) inhibition thus posing potential cardiotoxicity (Doggett et al. Reference Doggett, Ojo, Fan, Maly and Van Voorhis2014). Further extensive medicinal chemistry efforts finally led to the current lead TgCDPK1 inhibitor, (1-{4-amino-3-[2-(cyclopropyloxy)quinolin-6-yl]-1H-pyrazolo[3,4-d]pyrimidin-1-yl}-2-methylpropan-2-ol) that combines high activity and selectivity with favourable pharmacokinetic properties and low hERG activity (Vidadala et al. Reference Vidadala, Rivas, Ojo, Hulverson, Zambriski, Bruzual, Schultz, Huang, Zhang, Scheele, DeRocher, Choi, Barrett, Siddaramaiah, Hol, Fan, Merritt, Parsons, Freiberg, Marsh, Kempf, Carruthers, Isoherranen, Doggett, Van Voorhis and Maly2016). Note that the compound is bound to the protein via H-bonds of the pyrimidin ring to the main chain, while the hydrophobic cyclopropyloxy-quinoline moiety forms a large number of hydrophobic interactions (Fig. 5). Taken together, the structure-based approaches of drug development applied to TgCDPK1 has led to three different series of compounds with high inhibitory activity, good pharmacokinetic parameters and promising efficacy in murine models.

Fig. 3. Close-up of BKIs bound to TgCDPK1 in the ATP binding site. The gatekeeper residue Gly128 is depicted in magenta, key hydrophobic residue of the binding site are labelled and shown in grey (a) 1-(1-methylethyl)-3-(naphthalen-1-ylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine shown in ball-and-stick representation (PDB: 3i7b), (b) chemical structure of the ligand, (c) 1-tert-butyl-3-naphthalen-2-yl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PDB:3i7c) (Ojo et al. Reference Ojo, Larson, Keyloun, Castaneda, Derocher, Inampudi, Kim, Arakaki, Murphy, Zhang, Napuli, Maly, Verlinde, Buckner, Parsons, Hol, Merritt and Van Voorhis2010) and (d) chemical structure of the ligand. ATP, adenosine triphosphate; CDPKs, calcium-dependent protein kinases.

Fig. 4. The three different scaffolds for TgCDPK1 inhibitors (a) pyrazolpyrimidines, (b) acylbenzimidazoles and (c) 5-aminopyrazole-4-carboxamide. CDPKs, calcium-dependent protein kinases.

Fig. 5. Crystal structure of (1-{4-amino-3-[2-(cyclopropyloxy)quinolin-6-yl]-1H-pyrazolo[3,4-d]pyrimidin-1-yl}−2-methylpropan-2-ol) shown in stick representation bound to for TgCDPK1 shown in cartoon representation with selected residues depicted in sticks (Vidadala et al. Reference Vidadala, Rivas, Ojo, Hulverson, Zambriski, Bruzual, Schultz, Huang, Zhang, Scheele, DeRocher, Choi, Barrett, Siddaramaiah, Hol, Fan, Merritt, Parsons, Freiberg, Marsh, Kempf, Carruthers, Isoherranen, Doggett, Van Voorhis and Maly2016). CDPKs, calcium-dependent protein kinases.
CDPK1 INHIBITORS FOR RELATED PARASITES
Based on the success of developing specific TgCDPK1 inhibitors, recent work has branched out towards related apicomplexan parasites. For example, Neospora caninum, a cyst-forming parasite closely related to T. gondii represents the leading cause of abortion in cattle. This parasite expresses a CDPK1 with 96% sequence identity to TgCDPK1 where all residues in the active side are conserved, bar one conservative variation from phenylalanine to tyrosine (Ojo et al. Reference Ojo, Reid, Kallur Siddaramaiah, Muller, Winzer, Zhang, Keyloun, Vidadala, Merritt, Hol, Maly, Fan, Van Voorhis and Hemphill2014). Consequently, the crystal structures of TgCDPK1 and NcCDPK1 show very similar overall structures (root mean square deviations (rmsd) on C-alpha atoms 0·5 Å) and the same binding mode of a BKI. Importantly, a number of BKIs display comparable in-vivo activities (Ojo et al. Reference Ojo, Reid, Kallur Siddaramaiah, Muller, Winzer, Zhang, Keyloun, Vidadala, Merritt, Hol, Maly, Fan, Van Voorhis and Hemphill2014; Winzer et al. Reference Winzer, Müller, Aguado-Martinez, Rahman, Balmer, Manser, Ortega-Mora, Ojo, Fan, Maly, Van Voorhis and Hemphill2015). Members of the Cryptosporidium genus are the causative agent of cryptosporidiosis in immune-compromised patients and malnourished children (Shoultz et al. Reference Shoultz, de Hostos and Choy2016). CDPK1 from C. parvum Iowa II (CpCDPK1) shares a sequence identity of approximately 41% with TgCDPK1, however, the active site residues including the gatekeeper residue are highly conserved. The screening of BKI libraries resulted in highly active CpCDPK1 inhibitors based on the 5-aminopyrazole-4-carboxamide scaffold with clear potential for drug development (Castellanos-Gonzalez et al. Reference Castellanos-Gonzalez, Sparks, Nava, Huang, Zhang, Rivas, Hulverson, Barrett, Ojo, Fan, Van Voorhis and White2016). High-throughput screening for Plasmodium falciparum CDPK1 (PfCDPK1) inhibitors resulted in five chemical series, including the PP scaffold (Fig. 4a). PfCDPK1 shares a sequence identity of approximately 47% with TgCDPK1 and the gatekeeper residue threonine harbours a slightly larger side chain comparted to glycine. However, this side chain is still relatively small and mutational studies clearly indicated that these inhibitors bind at the same site (Ansell et al. Reference Ansell, Jones, Whalley, Hearn, Taylor, Patin, Chapman, Osborne, Wallace, Birchall, Large, Bouloc, Smiljanic-Hurley, Clough, Moon, Green and Holder2014). More recent studies with PfCDPK1 inhibitors based on the chemically very similar Imidazopyridazine scaffold appear to show that these compounds also target cyclic GMP dependent kinases as well as Heat Shock Protein 90. These findings question the prospect of PfCDPK1 inhibitors for further drug development (Green et al. Reference Green, Moon, Whalley, Bowyer, Wallace, Rochani, Nageshan, Howell, Grainger, Jones, Ansell, Chapman, Taylor, Osborne, Baker, Tatu and Holder2015). Taken together, these recent results show the potential of BKIs for future drug development in Toxoplasma and related parasites but they also illustrate the limitations of transferring detailed structural data to more distantly related proteins.
FUTURE CHALLENGES
Over the last 5 years there has been significant progress in the development of selective inhibitors of one of the key CDPKs from T. gondii achieved by taking advantage of a series of high-resolution crystal structures. While most of the previous research has focused on T. gondii, further efforts are currently underway to investigate inhibitors of CDPK1 from Cryptosporidium and Plasmodium spp. (Gaji et al. Reference Gaji, Checkley, Reese, Ferdig and Arrizabalaga2014; Green et al. Reference Green, Moon, Whalley, Bowyer, Wallace, Rochani, Nageshan, Howell, Grainger, Jones, Ansell, Chapman, Taylor, Osborne, Baker, Tatu and Holder2015; Crowther et al. Reference Crowther, Hillesland, Keyloun, Reid, Lafuente-Monasterio, Ghidelli-Disse, Leonard, He, Jones, Krahn, Mo, Dasari, Fox, Boesche, El Bakkouri, Rivas, Leroy, Hui, Drewes, Maly, Van Voorhis and Ojo2016). However, further research is required to unravel the biological roles of PfCDPKs and their potential as future drug targets (Kumar et al. Reference Kumar, Kumar, Ekka, Dvorin, Paul, Madugundu, Gilberger, Gowda, Duraisingh, Keshava Prasad and Sharma2017).
Although the most promising TgCDPK1 inhibitors show high efficacy in murine models, future research is required to increase solubility and bio-availability in order to proceed to clinical trials. Furthermore, current lead compounds only target the ATP binding site of TgCDPK1. However, allosteric kinase inhibitors and modulators have shown enormous potential to target specific kinases and could be further exploited (Fang et al. Reference Fang, Grutter and Rauh2013). Additional binding sites in less conserved regions such as the carbohydrate binding site recently discovered in TgCDPK2 can serve as starting points for the development of new inhibitors (Uboldi et al. Reference Uboldi, McCoy, Blume, Gerlic, Ferguson, Dagley, Beahan, Stapleton, Gooley, Bacic, Masters, Webb, McConville and Tonkin2015). Clearly, more works needs to be done to understand the role of the other members of the Apicomplexan CDPK family. In this regard, the recent development of CRISPR/Cas9 technology to modify the genes of members of the Apicomplexan family (Shen et al. Reference Shen, Brown, Lee and Sibley2014; Vinayak et al. Reference Vinayak, Pawlowic, Sateriale, Brooks, Studstill, Bar-Peled, Cipriano and Striepen2015) will greatly facilitate the detailed analysis of the biological function of CDPK family members (Long et al. Reference Long, Wang and Sibley2016; Wang et al. Reference Wang, Huang, Li, Chen, Ning and Zhu2016).
FINANCIAL SUPPORT
This work was generously supported by the BBSRC grant BB/M024156/1. In addition, we would like to thank the Biophysical Sciences Institute (BSI) for seed corn funding and the Institute for Advanced Studies (IAS) for a Senior Research Fellowship at Durham University to CLMV. EMC is grateful for a studentship by the Newcastle-Liverpool-Durham BBSRC Doctoral Training Partnership.







 Open access
Open access