


Introduction
In 2030 older people (individuals aged ≥60 years) are projected to account for almost 20 % of the global population(1). Despite this demographic shift in favour of older individuals, morbidity among this group is high(Reference Kingston, Robinson and Booth2). Many factors make an impact on older people’s health; however, an obesity pandemic is emerging as a significant global health concern(Reference Starr and Bales3–Reference Morgan, Mooney and Mc Auley7). Microcosmically, the UK illustrates the extent of the global obesity problem. Among females aged 65–74 years in the UK 30 % have a BMI ≥30 kg/m2, and are categorised as obese(Reference Baker8). The problem is even more pronounced among their male counterparts, as 33 % of males are categorised as obese within this age group(Reference Baker8). The problem extends to those aged ≥75 years, as 28 % of females are obese, while 23 % of males are obese in this age group. From a public health perspective these figures are alarming because obesity adversely affects several metabolic systems, and is synonymous with many conditions including cancer, type 2 diabetes mellitus (T2DM), hypertension and dyslipidaemia(Reference Pi-Sunyer9–Reference Reilly, Methven and McDowell11). However, due to its close association with overall cardiometabolic health, the impact that obesity has on cholesterol metabolism needs to be recognised(Reference Mc Auley, Gu and Dupre12,Reference Mc Auley and Mooney13) . The aim of the present review is to critically discuss the effects that obesity has on cholesterol metabolism and to reveal its significance for healthy ageing.
An overview of cholesterol metabolism
Fig. 1 outlines that cholesterol balance is maintained by the body responding to changes in ingestion, absorption, synthesis and excretion(Reference van der Wulp, Verkade and Groen14). Humans ingest a modest amount of dietary cholesterol (DC), which mixes with intestinal cholesterol(Reference Lu, Lee and Patel15). Absorption is controlled by cholesteryl ester (CE) hydrolase which liberates CE, facilitating the inclusion of free cholesterol (FC) into bile acid micelles(Reference Shamir, Johnson and Zolfaghari16). The intestinal protein Niemann–Pick C1-like 1 (NPC1L1) mediates cholesterol absorption into the enterocyte by clathrin-mediated endocytosis(Reference Jia, Betters and Yu17). ATP-binding cassette (ABC) transporters G5 and G8 (ABCG5/G8) control the efflux of cholesterol from the enterocytes to the lumen(Reference Li, Gu and Zhang18). Within the enterocyte, acetyl CoA acetyltransferase 2 (ACAT2) re-esterifies cholesterol(Reference Chang, Li and Chang19), which is combined with apoB-48, TAG and phospholipids, to generate a chylomicron(Reference Redgrave20). Upon entering the bloodstream, chylomicrons are acted on by lipoprotein lipase (LPL), which catalyses their TAG, liberating NEFA(Reference Mead, Irvine and Ramji21). Chylomicron remnants are removed from the circulation by hepatic remnant receptors(Reference Cooper22).
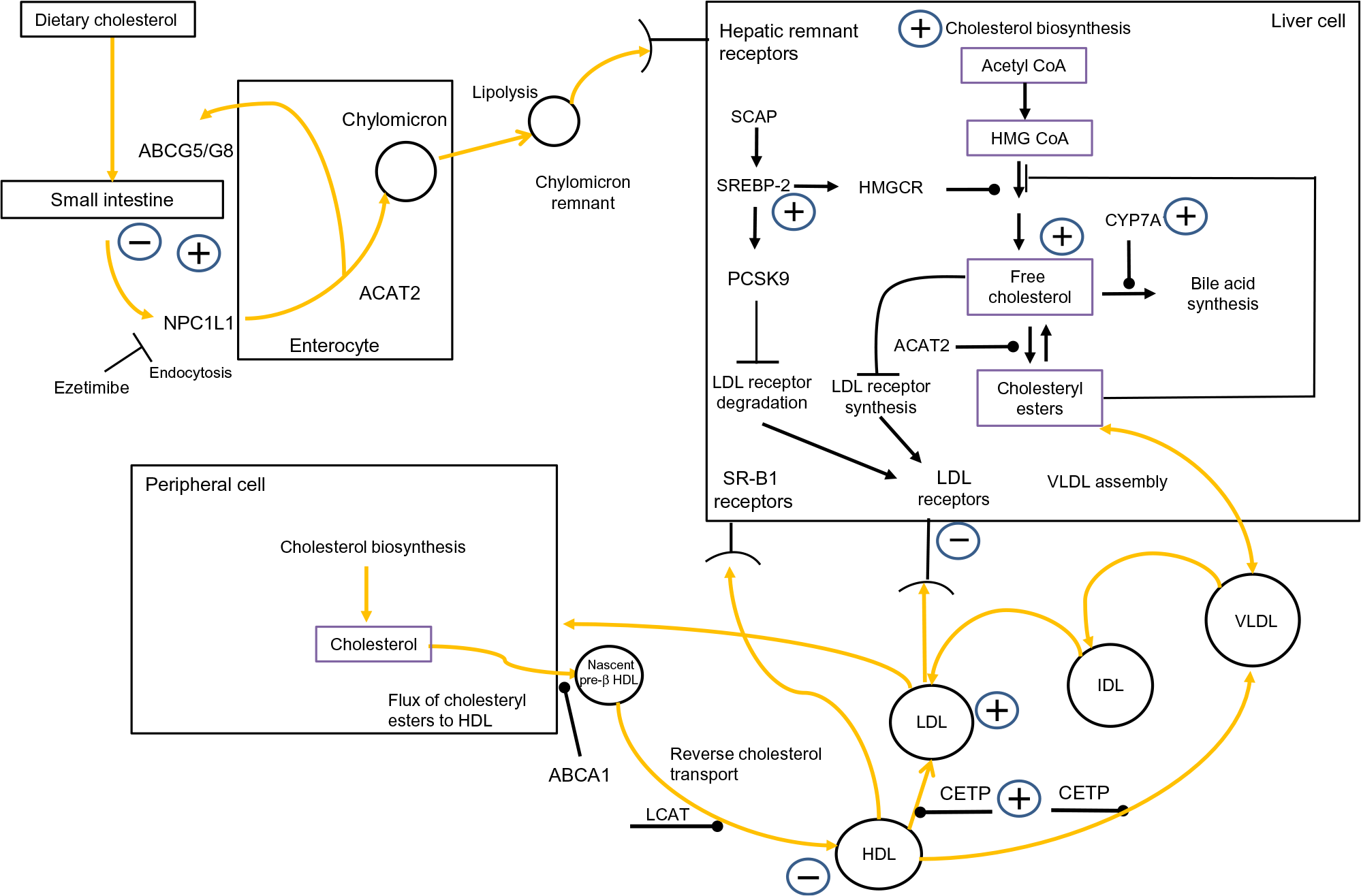
Fig. 1. Overview of whole-body cholesterol metabolism. Cholesterol metabolism is maintained by an array of regulatory processes which control absorption, synthesis, hepatic lipoprotein production, lipoprotein uptake and reverse cholesterol transport. The signs indicate where obesity has been shown to have an impact on cholesterol metabolism. Note: cholesterol absorption has both positive and negative signs associated with it, to indicate that certain studies have found that obesity decreases cholesterol absorption while other have found the opposite effect. ABCA1, ATP-binding cassette transporter; acetyl-CoA, acetyl coenzyme A; ABCG5/G8, ATP-binding cassette (ABC) transporters G5 and G8; ACAT2, acetyl CoA acetyltransferase 2; CETP, cholesteryl ester transfer protein; CYP7A1, cholesterol 7α-hydroxylase; IDL, intermediate-density lipoprotein; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGCR, HMG-CoA reductase; LCAT, lecithin–cholesterol acyltransferase; NPC1L1, Niemann–Pick C1-like 1; PCSK9, proprotein convertase subtilisin/kexin type 9; SCAP, sterol regulatory element-binding protein cleavage-activating protein; SREBP-2, sterol regulatory element-binding protein 2; SR-B1, scavenger receptor, class B type 1. For a colour figure, see the online version of the paper.
The liver is the main site of cholesterol synthesis(Reference DeBose-Boyd23), providing cholesterol and TAG for the assembly of VLDL(Reference Shelness and Sellers24). In the plasma, LPL hydrolyses VLDL to LDL, via intermediate-density lipoproteins(Reference Mendivil, Zheng and Furtado25). LDL-cholesterol (LDL-C) is removed by the LDL receptor (LDLr)(Reference Goldstein and Brown26) and LDLr-related protein 1(Reference van de Sluis, Wijers and Herz27). This process is governed by intracellular sterol levels(Reference Brown and Goldstein28). Increasing intracellular cholesterol activates insulin-induced genes (Insigs) proteins. Insig-1 and Insig-2 bind to sterol regulatory element-binding protein (SREBP) cleavage-activating protein (SCAP) in the endoplasmic reticulum, restricting the migration of the SCAP/SREBP complex to the Golgi(Reference Yabe, Brown and Goldstein29,Reference Yang, Espenshade and Wright30) . When sterol levels drop, SREBP-2 migrates to the Golgi where it is cleaved by subtilisin kexin isozyme/site-1 protease (SKI-1/S1P), and the intramembranous metalloprotease site-2 protease (S2P)(Reference Brown and Goldstein31). This releases the NH-terminal domain of SREBP-2 from the membrane. Two N-terminal fragments dimerise, then interact with importin-β, before entering the nucleus, to activate SREBP-2-regulated gene promoters(Reference Nagoshi and Yoneda32). A further regulatory point involves LDLr synthesis. Nuclear SREBP-2 increases the transcription of proprotein convertase subtilisin/kexin type 9 (PCSK9)(Reference Seidah, Awan and Chretien33). PCSK9 reduces the number of LDLr by increasing their metabolism, and subsequent degradation, restricting LDL uptake(Reference Seidah, Awan and Chretien33). High cellular cholesterol levels suppress SREBP-2 release from the endoplasmic reticulum, thus PCSK9 transcription is reduced, which subsequently increases LDLRr levels(Reference Lambert, Charlton and Rye34). The synchronised interplay of SREBP-2-induced transcription of both LDLr and PCSK9 regulates circulating LDL-C levels. Additionally, cholesterol entering a hepatic cell as part of LDL triggers ACAT2(Reference Goldstein and Brown26), which catalyses FC to CE(Reference Chang, Li and Chang19), and this cholesterol also activates cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme of bile acid synthesis(Reference Jackson, Ericsson and Edwards35).
By disrupting any of the mechanisms discussed above, obesity has the potential to provoke a rise in plasma LDL-C. Elevated LDL-C levels are inexorably linked to an increased risk of atherosclerotic CVD(Reference Sharrett, Ballantyne and Coady36–Reference Boekholdt, Arsenault and Mora38). Moreover, emerging evidence suggests that suboptimal LDL-C levels, in tandem with elevated serum uric acid, could present an increased risk of developing hypertension or the metabolic syndrome(Reference Cicero, Fogacci and Giovannini39,Reference Cicero and Fogacci40) .
A further important aspect of cholesterol metabolism is reverse cholesterol transport (RCT). RCT removes excess cholesterol from peripheral tissue(Reference Favari, Chroni and Tietge41). HDL are central to this. HDL ‘mop up’ excess cholesterol, generating HDL-cholesterol (HDL-C)(Reference Tuteja and Rader42). Central to RCT is the ferrying of FC and phospholipids to lipid-free apoA-I to form nascent pre-β HDL particles, in a process primarily regulated by ABCA1(Reference Oram and Lawn43,Reference Wang and Smith44) . Nascent HDL progress to mature HDL due to the esterification of cholesterol by lecithin–cholesterol acyltransferase (LCAT)(Reference Saeedi, Li and Frohlich45). Cholesterol within HDL can follow one of two routes to the liver. HDL can go directly to the liver and deposit their cholesterol by interacting with scavenger receptor class B, type 1 receptors(Reference Zhang, Da Silva and Reilly46). Second, cholesterol can be transferred to the liver via the action of CE transfer protein (CETP), which redistributes cholesterol to LDL and VLDL(Reference Fielding and Fielding47). Regardless of the route, HDL transfer cholesterol to the liver, where it can be effluxed directly as cholesterol or converted to bile salts(Reference Groen, Oude Elferink and Verkade48). It can then be excreted during enterohepatic circulation. Consequently, RCT is regarded as antiatherogenic(Reference Rye, Bursill and Lambert49); this is underscored by studies which have revealed an inverse relationship between HDL-C levels and the onset of premature CVD(Reference Castelli, Garrison and Wilson50,Reference Abbott, Wilson and Kannel51) . Intriguingly, HDL’s antiatherogenic role is thought to be enhanced further by possessing antioxidant properties(Reference Soran, Schofield and Durrington52). As with the mechanisms which regulate LDL-C levels, if obesity interferes with the processes underpinning RCT, this has the potential to modulate an individual’s risk of CVD.
Obesity and cholesterol metabolism
Cholesterol absorption
In pioneering work, Miettinen & Kesäniemi(Reference Miettinen and Kesäniemi53) identified a negative correlation between the fractional absorption of DC and obesity in middle-aged men; however, a mechanistic explanation for this result was not immediately apparent. In a follow-up investigation cholesterol absorption was also found to be inhibited in obese middle-aged males (BMI >31 kg/m2)(Reference Miettinen and Gylling54). On this occasion two explanations were proposed for this finding. It was posited that labelled DC could have contributed to sub-normal cholesterol absorption. Second, it was suggested that cholesterol absorption was inhibited by expanded biliary secretion. However, the precise reason why obesity inhibited cholesterol absorption remained unclear. More recent studies have added further intrigue to this puzzle. It has been found that treatment of obese hypercholesterolaemic subjects with the NPC1L1 inhibitor, ezetimibe, improved the lipid profile and insulin resistance (IR) in these subjects(Reference Adachi, Nakano and Yamamoto55,Reference Nakamura, Sato and Kanazawa56) . This suggests that obesity could in fact increase cholesterol absorption in obese subjects rather than inhibiting it, and an arbiter of this change could be NPC1L1. If obesity does increase cholesterol absorption, this effect could also be induced by provoking a rise in circulating bile acids. For example, Vincent et al. (Reference Vincent, Omar and Ghozlan57) found that the postprandial bile acid response is increased in obese male and female patients with T2DM compared with age-matched normoglycaemic individuals. Further evidence that obesity influences bile acid metabolism comes from studies of the gut microbiome(Reference Foley, O’Flaherty and Barrangou58). For instance, in one study it was found that bile salt hydrolase is the arbiter of host–microbiome interactions which modulated weight gain, and lipid metabolism in a murine model(Reference Joyce, MacSharry and Casey59). Specifically, the expression of cloned bile salt hydrolase enzymes in the gastrointestinal tract of gnotobiotic or conventionally raised mice significantly modified plasma bile acid signatures and regulated the transcription of important genes involved in cholesterol metabolism (Abcg5/8) both hepatically and intestinally. Moreover, high-level expression of bile salt hydrolase in conventionally raised mice resulted in a significant drop in host weight gain, plasma cholesterol levels, and hepatic TAG. As an adjunct to this finding it has been shown that the farnesoid X receptor (FXR) has a central role to play in modulating host–microbiome dialogue. For instance, mouse models of diet-induced obesity have shown that both the microbiome and FXR signalling are required for weight gain(Reference Parseus, Sommer and Sommer60,Reference Li, Jiang and Krausz61) .
Cholesterol synthesis
Cholesterol synthesis is also affected by obesity; sterol-balance studies have shown that increased weight gain results in a higher rate of cholesterol synthesis(Reference Miettinen62,Reference Nestel, Schreibman and Ahrens63) . It is uncertain how weight gain induces a higher rate of cholesterol synthesis. However, it is important to acknowledge that hepatic HMGCR increases in obese subjects(Reference Angelin, Backman and Einarsson64). This would naturally result in an increase in hepatic cholesterol production, something which has been observed in obese subjects(Reference Stahlberg, Rudling and Angelin65). For example, in a study involving seventeen morbidly obese middle-aged males, it was found that the activity, and mRNA levels of HMGCR, was higher in the obese subjects, when compared with a lean control group(Reference Mamo, Watts and Barrett66). Moreover, the activity and mRNA level of cholesterol 7α-hydroxylase (CYP7A1) also increased compared with controls. The activity of ACAT2, and LDLr mRNA levels were elevated in these subjects. Such alterations have the potential to make an impact on the normal functioning of hepatic LDLr. For example, it was also found in this study that the binding of LDL to the LDLr was reduced by 50 % when compared with controls(Reference Mamo, Watts and Barrett66).
Hepatic free cholesterol/bile acid accumulation and non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) encompasses a number of hepatic pathologies, from fatty liver disease to non-alcoholic steatohepatitis (NASH), a condition which can progress to cirrhosis(Reference Farrell and Larter67–Reference Moore71). NAFLD has a higher occurrence in males than females, and its prevalence increases with age(Reference Wang, Xu and Peng72,Reference Bertolotti, Lonardo and Mussi73) . NAFLD is closely associated with IR and hyperinsulinaemia and is prevalent in 70–80 % of obese individuals(Reference Loomba, Abraham and Unalp74). Moreover, it has recently been associated with key parameters of cardiovascular health, including arterial stiffness(Reference Cicero, Gitto and Fogacci75). Traditionally, NAFLD has been associated with increased hepatic TAG; however, in recent years there has been growing evidence linking altered cholesterol metabolism with the aetiology of NAFLD(Reference Arguello, Balboa and Arrese76–Reference Tirosh78). For instance, hepatic FC accumulates in obese diabetic mice and results in steatohepatitis(Reference Van Rooyen, Larter and Haigh79). Most recently in mice it has been found that hepatic cholesterol, but not hepatic TAG, increased with age(Reference Seo, Kang and Choi80). Focusing on human subjects, the intake of high levels of DC has been associated with NASH(Reference Musso, Gambino and De Michieli81,Reference Ioannou, Morrow and Connole82) . Mechanistically, it would appear that cholesterol synthesis is up-regulated in NAFLD. This was demonstrated by a study which examined the expression of an array of genes associated with cholesterol metabolism(Reference Min, Kapoor and Fuchs83). In the investigation, twenty middle-aged subjects with NAFLD (mean BMI 34·1 kg/m2) and NASH (mean BMI 34·2 kg/m2) were compared with twenty obese controls (33·2 kg/m2) and six lean normal controls (mean BMI 21·4 kg/m2). It was found that NAFLD was associated with increased SREBP-2 maturation, HMGCR expression and decreased phosphorylation of HMGCR. Additionally, CE hydrolase was increased, while ACAT2 remained unchanged. Moreover, LDLr expression decreased significantly. Also, HMGCR expression was correlated with FC, histologic severity of NAFLD and LDL-C levels. Bile acid homeostasis is also affected as a result of NAFLD(Reference Claudel, Staels and Kuipers84). Individuals with NASH have elevated levels of bile acids which can accumulate hepatically(Reference Puri, Daita and Joyce85). Bile acid signalling is regulated by the FXR, which contributes to overall cholesterol metabolism(Reference Claudel, Staels and Kuipers84). Interestingly, it has been shown in mice that the gut microbiota can modulate obesity via this receptor. The gut microbiota promoted weight gain and hepatic steatosis in an FXR-dependent manner between Fxr–/– and wild-type mice(Reference Parseus, Sommer and Sommer60). Moreover, bile acid profiles and the composition of faecal microbiota differed between Fxr–/– and wild-type mice. The finding that the gut microbiota induced liver steatosis in an FXR-dependent manner was suggested to be due to increased expression of CD36, apoC2 and VLDL receptor, all of which are involved in lipoprotein uptake. Thus, increased steatosis was thought to be attributed to the augmented expression of lipogenic genes or diminished expression of genes associated with fatty acid oxidation. In terms of promoting obesity mechanistically the authors showed that the gut microbiota of Fxr-deficient mice was defined by a phylum-wide rise in Bacteroidetes and phylum-wide reduction of Firmicutes. Thus, the gut microbiota changes in response to diet and is associated with an obesity phenotype, which is mediated by FXR signalling.
Lipoprotein dynamics and reverse cholesterol transport
Lipoprotein processing is significantly impaired due to obesity. Morbidly obese middle-aged patients have been found to have lower expression of LPL and LDLr-related protein 1 in their visceral tissue(Reference Clemente-Postigo, Queipo-Ortuno and Fernandez-Garcia86). LPL expression was also lower in the subcutaneous adipose tissue of these subjects. The decrease in the expression of LDLr-related protein 1 is probably a contributing factor to the increase in plasma LDL-C, which often, but not always, accompanies obesity(Reference Kesaniemi and Grundy87). More strikingly, obesity has regularly been associated with an increase in atherogenic small dense LDL(Reference Gerber, Nikolic and Rizzo88–Reference Meyer, Stewart and Brown90). Obesity also lowers HDL-C regardless of age, sex or ethnic background, while an inverse association between HDL-C levels and BMI has also been observed(Reference Denke, Sempos and Grundy91–Reference Sternfeld, Sidney and Jacobs93). Intriguingly, in the Framingham Offspring Study, the effect of increased BMI on total cholesterol and LDL-C was not as strong as it was for HDL-C(Reference Lamon-Fava, Wilson and Schaefer94). More recently, an inverse association has been found between LDL-C and BMI in morbidly obese subjects(Reference Shamai, Lurix and Shen95). Low levels of the major apolipoprotein component of HDL (apoA-I) have also been found to be associated with obesity in the Framingham Offspring Study in men and women(Reference Garrison, Wilson and Castelli96). Moreover, it has been revealed in a mouse model that increased apoA-I could have an anti-obesity effect. Increased energy expenditure and up-regulation of uncoupling protein 1 in brown fat were associated with high levels of apoA-I(Reference Ruan, Li and Zhang97). In addition, other apolipoproteins have been shown to be influenced by obesity. Elevated levels of fasting and postprandial apoB-48 have been measured in obese human subjects(Reference Mamo, Watts and Barrett66). Obesity could also have an impact on the functional capacity of HDL-C. For example, the antioxidant capacity of HDL appears to be compromised in obese subjects(Reference Sorrentino, Besler and Rohrer98). Obesity interferes with other components of RCT, for example, obesity causes impairment in RCT due to reduced plasma cholesterol uptake and efflux by hepatocytes and adipocytes in ob/ob mice(Reference Duong, Uno and Nankivell99). Plasma CETP levels have also been positively correlated with obesity(Reference Arai, Yamashita and Hirano100). The likely reason for this finding is that adipose tissue is a significant source of CETP(Reference Radeau, Lau and Robb101). Intriguingly, LCAT deficiency has been suggested to confer a degree of protection from the development of obesity. Tentative evidence for this finding comes from a study of LCAT-null mice which appeared to be protected from diet-induced obesity(Reference Li, Hossain and Sadat102).
Commonality between obesity and ageing
It is possible that obesity superimposed on ageing could expedite the age-associated dysregulation of cholesterol metabolism(Reference Morgan, Mooney and Wilkinson103–Reference Mc Auley and Mooney105). Taking LDL-C as an example, increasing age is associated with a rise in LDL-C in males and females, in both cross-sectional(Reference Carroll, Lacher and Sorlie106,Reference Anderson, Wilson and Garrison107) and prospective studies(Reference Criqui, Frankville and Barrett-Connor108,Reference Ettinger, Wahl and Kuller109) . It is not known how ageing contributes to a rise in LDL-C; however, there are solid reasons to believe that a decline in the hepatic clearance rate of LDL-C is a factor(Reference Morgan, Mooney and Wilkinson110,Reference Mc Auley, Wilkinson and Jones111) . For example, human ageing is associated with a drop in the number of hepatic LDLr(Reference Lee, Paz and Gallop112). Obesity can also results in a rise in LDL-C(Reference Kesaniemi and Grundy87,Reference Lamon-Fava, Wilson and Schaefer94) , although in some observational studies it is only weakly associated(Reference Aronne, Brown and Isoldi113,Reference Kannel, Gordon and Castelli114) , or not associated at all(Reference Laclaustra, Lopez-Garcia and Civeira115). An obesity-induced rise in LDL-C could have the same mechanistic underpinning as ageing, because it is also associated with a decline in hepatic LDLr numbers. Also similar to obesity, HDL-C levels decrease with age in human subjects in certain studies(Reference Cooney, Dudina and De Bacquer116). For instance, HDL-C levels have been observed to decrease with age in both men and women in prospective studies(Reference Ettinger, Wahl and Kuller109,Reference Heiss, Tamir and Davis117) . Although HDL-C levels do not change with age in most cross-sectional studies(Reference Flynn, Nolph and Baker118,Reference Frishman, Ooi and Derman119) , they have been shown to decrease in others(Reference Ferrara, Barrett-Connor and Shan120). A decline in HDL-C with age could be due to disrupted CETP activity. If this is the case it would be mechanistically similar to the putative effect that obesity has on RCT(Reference Arai, Yamashita and Hirano100). Ageing also makes an impact on lipoprotein processing by reducing the activity of plasma LPL by as much as 55–60 %(Reference Niemi and Nikkila121,Reference Brodows and Campbell122) . This is similar to the metabolic affect that obesity has on LPL(Reference Wang and Eckel123). In rodents it has been found that bile acid synthesis diminishes with age(Reference Bertolotti, Gabbi and Anzivino124). This is also similar to obesity because in men and women a decrease in the conversion of cholesterol to bile acids has been identified in certain studies(Reference Welsh, Sharma and Abramson125,Reference Schulze, Manson and Ludwig126) . However, other studies conflict with this and suggest that obesity results in an increase in bile acid synthesis(Reference Haeusler, Camastra and Nannipieri127).
The landscape of an obese state also resonates with the free radical theory of ageing(Reference Harman128). For example, obesity is associated with oxidative stress via increased generation of reactive oxygen species (ROS)(Reference Fernandez-Sanchez, Madrigal-Santillan and Bautista129). As visceral fat stores increase, adipocytes generate increasing levels of ROS(Reference Fujita, Nishizawa and Funahashi130). Consequently, oxidative stress results in IR within adipose and peripheral tissue(Reference Keaney, Larson and Vasan131). It has been suggested that high levels of ROS impinge on intracellular cholesterol homeostasis(Reference Mc Auley and Mooney132). ROS have been found to up-regulate the activity of hepatic HMGCR in rodent hepatic tissue, leading to an increase in cholesterol synthesis(Reference Seo, Kang and Choi80,Reference Pallottini, Martini and Bassi133–Reference Pallottini, Martini and Cavallini135) . ROS are also implicated in the pathogenesis of atherosclerosis, where oxidation of LDL is regarded as a key event in the initial stages of atherosclerosis formation(Reference Singh, Mengi and Xu136). Despite the many parallels between obesity and ageing, a number of differences do exist. In contrast to obesity it has been revealed that cholesterol absorption efficiently increases with age; however, there is a paucity of evidence for this in human subjects and findings are confined to rodent studies(Reference Hollander and Morgan137). Mechanistically it appears that ageing suppresses the expression of Abcg5 and Abcg8, and up-regulates the expression of Npc1l1 in murine models(Reference Duan, Wang and Ohashi138). In rodents it has been found that bile acid synthesis declines with age(Reference Bertolotti, Gabbi and Anzivino124). This contrasts with obesity. Also, unlike obesity, it has been found that hepatic ACAT2 activity decreases with age in Watanabe heritable hyperlipidaemic rabbits(Reference Shiomi, Ito and Fujioka139).
Diet: a key modulator of obesity and cholesterol metabolism
An obese state is the result of an imbalance between the amount of energy consumed by an individual and the amount of energy they expend(Reference Camacho and Ruppel140). Moreover, it is generally regarded that excess consumption of dietary fat plays a role in the development of obesity(Reference Bray and Popkin141). Taking this a step further, a clear link between diet, cholesterol metabolism and obesity centres on the excessive intake of DC. For instance, DC has been shown to exacerbate hepatic steatosis and inflammation in obese LDLr-deficient mice(Reference Subramanian, Goodspeed and Wang142). Moreover, in this study, the consumption of DC exacerbated hepatic macrophage infiltration, apoptosis and oxidative stress. Excessive intake of DC has also been shown to result in the accumulation of hepatic cholesterol in obese diabetic mice(Reference Van Rooyen, Larter and Haigh79). The accumulation of cholesterol was attributed to changes in some of the regulator mechanisms discussed previously, including the up-regulation of LDLr, via activation of SREBP-2, a drop in the conversion of cholesterol to bile acids, and suppression of bile acid excretion in bile.
In addition to the intake of fat/DC, high intakes of dietary sugars have been associated with obesity and unfavourable lipid levels in both men and women(Reference Welsh, Sharma and Abramson125,Reference Schulze, Manson and Ludwig126) . In particular, dietary fructose has emerged as an important dietary factor which contributes to the hepatic dysregulation of cholesterol metabolism(Reference Moore, Gunn and Fielding69,Reference Campos and Tappy143) . For instance, high fructose consumption increases serum PCSK9 concentrations and reduces liver LDLr protein levels in hyperlipidaemic hamsters(Reference Dong, Singh and Azhar144). In human subjects, it has been found that the consumption of fructose and high-fructose maize syrup increases LDL-C, and apo-B in both men and women(Reference Stanhope, Bremer and Medici145). Moreover, NASH is associated with animal models fed a high-fat, high-fructose diet(Reference Kohli, Kirby and Xanthakos146,Reference Zhang, Han and Man147) . Consumption of excessive dietary fructose has also been associated with cognitive decline in older adults(Reference Lakhan and Kirchgessner148). This is intriguing because obesity can be correlated with poor cognitive performance in older adults(Reference Goncalves Damascena, Batisti Ferreira and Dos Santos Teixeira149). Taking this a step further, it is possible that fructose makes a mechanistic contribution to the pathogenesis of Alzheimer’s disease by interfering with lipid metabolism. For example, animal models of dementia suggest that excessive consumption of fructose induces IR and promotes dementia pathogenesis(Reference Cao, Lu and Lewis150–Reference Cisternas, Salazar and Serrano152). This is thought to occur as follows: IR is associated with elevated plasma ceramides which interfere with lipoprotein metabolism and amyloidogenic processing, resulting in the deposition of β-amyloid peptides, which is a hallmark of Alzheimer’s disease(Reference Martins and Creegan153).
In certain circumstances age could potentially offer a degree of protection against diet-induced obesity. In a recent study which compared the response of young and old mice to a Western diet, it was found that old mice did not show a higher body weight or adipose tissue mass, when compared with their young counterparts(Reference Vercalsteren, Vranckx and Frederix154). Significantly, and of direct relevance to the underlying hepatic health of older individuals, it was found that the aged mice did have a build up of hepatic lipid on the Western diet. As well as the type and amounts of nutrients consumed, it has been found that meal frequency, timing and regularity are also associated with obesity(Reference Maugeri, Kunzova and Medina-Inojosa155). Recently this has been shown to have important implications for cholesterol metabolism. In a cross-sectional study of non-institutionalised and non-pregnant healthy Taiwanese adults (aged ≥19 years old), it was found that higher energy intake at night time is associated with elevated total cholesterol and LDL-C levels(Reference Chen, Chuang and Chang156). This was an interesting finding, although a mechanistic explanation for this discovery was not posited. Regardless, the study presents the possibility that meal timing and frequency could make an impact on both obesity and cholesterol metabolism, which is an intriguing prospect.
Alcohol consumption also affects cholesterol metabolism. Evidence suggests that moderate to low alcohol consumption increases levels of HDL-C and decreases levels of LDL-C(Reference Brien, Ronksley and Turner157). However, it is important to be cautious as alcohol has a limited therapeutic range and only modest drinking appears to be beneficial. This is partly due to the energy richness of alcohol, as its excessive consumption can result in increased weight gain in certain individuals(Reference Traversy and Chaput158). Moreover, chronic alcohol intake affects lipid metabolism broadly by provoking the increased synthesis of TAG and subsequent hypertriacylglycerolaemia(Reference Bessembinders, Wielders and van de Wiel159). In addition, it has been observed that alcohol consumption decreases the activity of LPL, which is also associated with hypertriacylglycerolaemia(Reference Pownall160). From the perspective of HDL metabolism, in individuals with alcohol dependence syndrome no association between plasma HDL-C and the number of drinks consumed per d has been observed in these individuals, indicating that only low-level consumption of alcohol is beneficial(Reference Bell, Stromme and Steensland161). Furthermore, in a longitudinal study of alcohol consumption, the long-term effect of total alcohol consumption on the change in HDL-C was observed to be a non-linear relationship(Reference Huang, Li and Shearer162). The mechanistic explanation for this is thought to centre on the pathophysiological effect of alcohol on the liver which results in a decrease in the hepatic production of HDL in these subjects(Reference Devenyi, Robinson and Kapur163).
Alarmingly, the excessive intake of alcohol has been increasing among older individuals in certain populations(Reference Almeida, McCaul and Hankey164). In a recent study involving a cohort of Australian males (aged ≥ 65 years), which explored the association between alcohol intake and body composition, it was found that participants who consumed ≥5 alcoholic drinks/d had a greater BMI, fat mass index, waist circumference, percentage body fat and lower lean mass than non-drinkers(Reference Coulson, Williams and Brennan165). This has metabolic consequences for older individuals because alcohol consumption augments lipid synthesis via sterol SREBP-1(Reference You, Fischer and Deeg166). This is possibly mediated by acetaldehyde, which contributes to an increase in the synthesis of SREBP-1, which in turn augments cholesterol and fat synthesis(Reference You, Fischer and Deeg166). Alcohol consumption has also been shown to dysregulate hepatic fatty acid oxidation(Reference Rogers, Ajmo and You167) and decrease the secretion of VLDL(Reference Kharbanda, Todero and Ward168). A drop in VLDL secretion could result in a decrease in the conversion of VLDL-C to LDL-C, and be responsible for the decrease in LDL-C associated with low to moderate alcohol intake(Reference Nakanishi, Yoshida and Nakamura169). Furthermore, this mechanism could explain the decreased risk of alcohol dependence with increased LDL-C levels identified among some of the participants of a recent case–control study investigating alcohol consumption and obesity(Reference Goodyear, Lee and Schwandt170).
On the flip side of the coin, emerging research has revealed that certain novel dietary components improve both cholesterol metabolism and have anti-obesity effects. For example, a diet high in fruit and vegetables is associated with a lower risk of obesity/body adiposity(Reference Rautiainen, Wang and Lee171–Reference Yu, DeClercq and Cui173). Moreover, certain components of fruits and vegetables have a favourable effect on cholesterol metabolism. For instance, soluble fibre exerts a favourable effect by decreasing LDL-C levels(Reference Bazzano174). It has been found that 3 g/d of soluble fibre can lower total cholesterol and LDL-C by about 0·13 mmol/l(Reference Brown, Rosner and Willett175). Several mechanisms have been suggested to account for this effect, including the inhibition of bile salt intestinal re-absorption, and a diminished glycaemic response, which results in a drop in insulin-stimulated hepatic cholesterol synthesis(Reference Gunness and Gidley176). The gut is also thought to be the site of action of plant sterols. Plant sterols are naturally occurring compounds found in fruit and vegetables which are structurally related to cholesterol, differing only in the structure of their side chains(Reference He, Zhu and Chen177). Consumption of 1·8–2·0 g/d of plant sterols has been shown to lower both total cholesterol and LDL-C concentrations by 10–15 % in different population groups(Reference Katan, Grundy and Jones178,Reference Trautwein, Vermeer and Hiemstra179) . Although the precise mechanism by which LDL-C is lowered is uncertain, it is generally regarded that plant sterols inhibit intestinal cholesterol absorption(Reference Plat, Baumgartner and Vanmierlo180). Not only have plant sterols been associated with decreased LDL-C. More recently, the consumption of high levels of phytosterols, which includes plants sterols, have been associated with decreased rates of obesity. For example, a recent cross-sectional study of Chinese adults (aged 18–60 years) revealed that higher consumption of phytosterols was associated with lower BMI, waist circumference, and a lower prevalence of overweight/obesity/abdominal obesity in this population group(Reference Li, Li and Li181). Intriguingly the administration of both phytosterols and red rice was recently studied in mildly hypercholesterolaemic subjects(Reference Cicero, Fogacci and Rosticci182). In tandem these two nutraceuticals had a more significant impact on LDL-C levels, when compared with either phytosterols or red rice on their own. Diet also has an important role to play in terms of alleviating the metabolic consequence of obesity. Most recently, it has been shown that medium-chain SFA could illicit a degree of protection against obesity-induced co-morbidities such as diabetes in obesogenic mice(Reference Zacek, Bukowski and Mehus183). Moreover, in a rat model it has been found that medium-chain SFA could help prevent NAFLD(Reference Ronis, Baumgardner and Sharma184). Mechanistically, it is thought the beneficial effects of medium-chain SFA consumption could be induced via their preferential β-oxidation over long-chain SFA(Reference St-Onge and Jones185). A further way that diet has been suggested to provide a means of treating obesity is by modulating the gut microbiome. For instance, in an obese population which adhered to a Mediterranean diet for 1 year, it was found that the Mediterranean diet exerted a protective effect against T2DM development by modulating specific changes in the gut microbiota. Specifically, this involved increasing the abundance of Faecalibacterium prausnitzii and Roseburia species.
Cholesterol metabolism and obesity in older individuals
Certain individuals have a metabolic profile which does not appear to be overtly affected by obesity. Such individuals are known as ‘metabolically healthy obese’(Reference Phillips186). In a study which investigated obesity in the US population, it was found that 31·7 % of obese adults (about 19·5 million) were metabolically healthy obese(Reference Wildman, Muntner and Reynolds187). The prevalence of metabolically healthy older individuals was 14·3 % among those aged 65–79 years, and 22·1 % among those aged ≥80 years. Healthy participants were defined by having 0 or 1 cardiometabolic abnormality. Unhealthy individuals were defined as having ≥2 cardiometabolic abnormalities. Among individuals with ≥2 metabolic abnormalities, the two most common cardiometabolic risk factor combinations were high TAG level/low HDL-C level and high blood pressure/high glucose level. In a similar investigation it was found that among participants with ≤1 metabolic abnormality, obesity was associated with a greater risk of developing multiple metabolic abnormalities(Reference Achilike, Hazuda and Fowler188). Significantly, lipid metabolism was also key, as TAG and HDL-C levels predicted an individual’s progression to a metabolically unhealthy obese state. Within obesity research, a further puzzle exists; there are situations where being overweight/mildly obese appears to be beneficial. This is known as the ‘obesity paradox’(Reference Banack and Kaufman189). In an ageing context, the obesity paradox appears to confer a survival advantage in older patients (generally those aged >50 years) who have conditions such as CVD, arthritis and kidney disease(Reference Lavie, De Schutter and Milani190). Focusing specifically on cholesterol metabolism, and its intersection with the obesity paradox, normal total serum cholesterol levels have been reported in morbidly obese individuals. In a study of 3312 women (aged ≥ 18 years) it was found that the percentage of individuals with normal total serum cholesterol levels (<200 mg/dl; <5·18 mmol/l) decreased with increasing BMI, from 55 % in those with a BMI <20 kg/m2 to 28 % in women with a BMI of 30–35 kg/m2(Reference Vierhapper, Nardi and Grosser191). Total serum cholesterol >7·75 mmol/l was found in 2 % of individuals with a BMI <20 kg/m2, but in 6 % of the group with a BMI between 30 and 35 kg/m2. Among morbidly obese women (BMI >40 kg/m2), 39 % had total serum cholesterol levels <5 mmol/l. Thus, it would appear in morbidly obese women that there is a significant number of individuals with normal total serum cholesterol levels. Such findings were also identified in a study which examined individuals (aged 20–64 years) with a BMI in the range 34–77 kg/m2(Reference Dixon and O’Brien192). It was found that mean total cholesterol levels in the obese group fell with increasing BMI. Moreover, LDL-C levels were lower in obese men (3·65 mmol/l v. the control group, 4·17 mmol/l).
The association between obesity and cholesterol metabolism has been studied to a limited extent in the oldest old (individuals aged ≥80 years). Despite this, the oldest old who are obese have a higher prevalence of morbidity(Reference Da Cruz, Almeida and Schwanke193). In a study which examined the oldest old among a group aged 60–85 years, it was reported that obesity was associated with shorter survival plus a higher incidence of CHD and T2DM(Reference Bowman, Delgado and Henley194). When cholesterol metabolism has been examined in the oldest old some intriguing findings have been revealed. In the Leiden 85-Plus Study it was observed that both high and low levels of LDL-C had a similar impact on mortality risk(Reference Weverling-Rijnsburger, Jonkers and van Exel195). Interestingly, this finding occurred despite CVD being the main cause of mortality in these subjects. Similar observations have been identified in several other studies which have examined the lipoprotein profile of the oldest old(Reference Ravnskov, Diamond and Hama196,Reference Al-Mallah, Hatahet and Cavalcante197) . Interestingly, during a 3-year follow-up study involving the Chinese oldest old it was found that for each 1 mmol/l increase of LDL-C concentration there was a corresponding 19 % decrease in 3-year all-cause mortality(Reference Lv, Yin and Chei198). These findings are intriguing and require a biological explanation. The oldest old in general are in a state of multi-morbidity(Reference Formiga, Ferrer and Sanz199). Based on this premise it is logical that low levels of LDL-C could be one particular clinical manifestation of underlying multi-morbidity. Ageing superimposed on cholesterol metabolism in a metabolically unhealthy obese individual or a metabolically unhealthy normal-weight individual could theoretically contribute to a drop in LDL-C. The conceptual framework outlined in Fig. 2 suggests that an obese state/poor metabolic health combined with an age-associated rise in hepatic ROS levels results in a rise in HMGCR activity(Reference Dai, Chiao and Marcinek200,Reference Mc Auley201) . In a normolipidaemic individual this would result in a rise in LDL-C due to the homeostatic down-regulation of LDLr synthesis. However, if there is also an age-associated decrease in ACAT2 activity, this reduces the conversion of FC to CE. Consequently, VLDL-C production would drop and there would be a concomitant reduction in LDL-C. As intracellular levels of FC accumulate and oxidative stress progresses, this state could advance to NAFLD(Reference Zhao, Chen and Tang202). As there is a strong association between NAFLD(Reference Francque, van der Graaff and Kwanten203) and CVD(Reference Dulai, Singh and Patel204), this in theory could increase an older individual’s risk of mortality, and help to account for the association between low levels of LDL-C and increased risk of mortality, which has been observed in certain studies involving the oldest old.
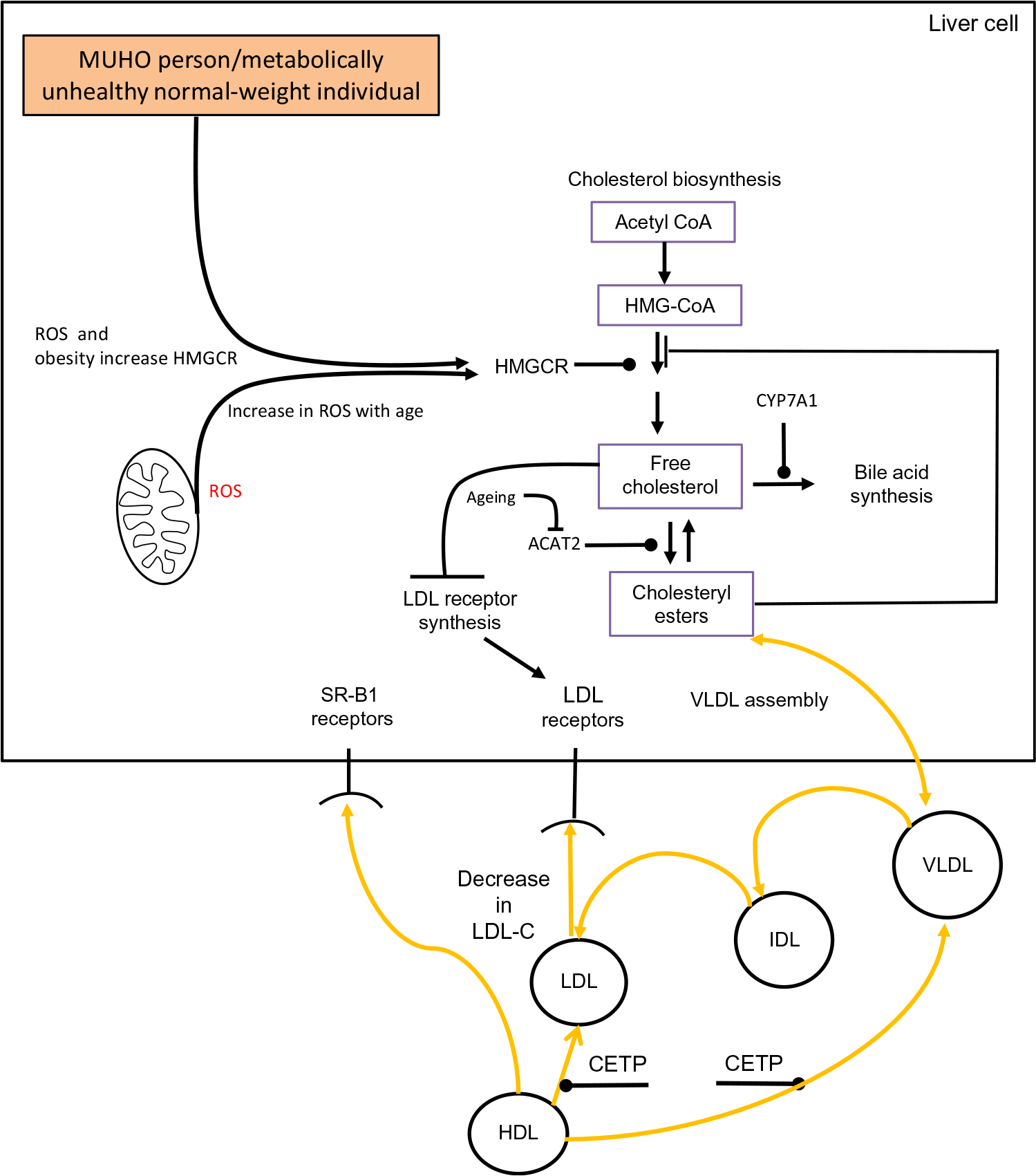
Fig. 2. Conceptual model of obesity/ageing-induced changes to hepatic cholesterol metabolism which could result in low levels of LDL-cholesterol (LDL-C), a metabolic feature which has been observed in a number of studies involving the oldest old. Acetyl-CoA, acetyl coenzyme A; ACAT2, acetyl CoA acetyltransferase 2; CETP, cholesteryl ester transfer protein; CYP7A1, cholesterol 7α-hydroxylase; IDL, intermediate-density lipoprotein; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGCR, HMG-CoA reductase; MUHO, metabolically unhealthy obese; ROS, reactive oxygen species; SR-B1, scavenger receptor, class B type 1. For a colour figure, see the online version of the paper.
Conclusions
Obesity among older individuals has increased significantly. The present review has revealed that obesity has a pleiotropic effect on cholesterol metabolism. Obesity affects cholesterol absorption, synthesis, lipoprotein processing, and results in the accumulation of cholesterol hepatically. Many of the changes are similar to how ageing intersects with cholesterol metabolism, and it can be suggested that an obese state superimposed on ageing has the potential to exacerbate the dysregulation of cholesterol metabolism, which occurs with advancing age. The present review also revealed diet as a key factor which links an obese state to important changes which occur in hepatic cholesterol metabolism. In particular, the excessive intakes of dietary lipids and fructose were highlighted as key factors which underpin conditions such as NAFLD. Careful attention needs to be placed on this association. This review also highlighted a number of anomalies which exist in this field. Firstly, there are certain individuals who, despite being in an obese state, appear to be normolipidaemic, and it is not immediately clear why this is the case. The second anomaly centres on the oldest old: in particular, the association between low levels of LDL-C and an increased risk of mortality, which has been observed in a number of studies. A tentative explanation for this association was presented, which centred on obesity/an unhealthy metabolic state and its intersection with ageing as important factors underpinning this anomaly. However, this is only one possible explanation and to fully elucidate this intriguing anomaly, it is necessary for the dynamics of cholesterol metabolism in the oldest old to be investigated to a much greater extent. To date there has been a paucity of research in this area. Finally, the present review has raised a broader question which relates to the public health challenge surrounding an ageing global population which is becoming increasingly obese. There is no straightforward solution to this problem. However, one possible strategy could involve adopting public health initiatives which target middle-aged individuals and educating them about the deleterious health implications of being overweight/obese. Increased awareness among this group could lead to better health in later life. To this end it is vital that public health interventions are initiated which make both younger and middle-aged individuals cognisant of appropriate lifestyle choices which optimise their chances of growing old healthily. If this issue is not addressed in coming years, more and more people will reach old age in poor metabolic heath due to being obese.
Acknowledgements
I would like to thank Dr Amy Morgan for proof reading the document.
There are no conflicts of interest.


