I. INTRODUCTION
Water splitting is a promising approach to transform solar energy and to store it in the form of green chemicals or fuels, such as hydrogen. Reference Gray1–Reference Turner6 As one of the half reactions, the oxygen evolution reaction (OER) is the key step in providing the reductive equivalents and protons for performing reduction of proton to hydrogen. Reference Zhong and Gamelin7,Reference Cao, Lai and Du8 However, the water oxidation reaction is the bottleneck step, due to the formation of the O–O bond, which typically leads to slow kinetics and high overpotentials. Reference Barber9 Thus, there is a high demand for an electrocatalyst with high activity, excellent stability, and low overpotential for OER. The materials based on precious metals, such as IrO2 and RuO2, have been extensively studied as water oxidation catalysts since the late 1970s, but the low abundance and high-cost of these noble metals significantly hamper their practical applications. Reference Harriman, Pickering, Thomas and Christensen10–Reference Carmo, Fritz, Merge and Stolten12 Therefore, recent efforts have been dedicated to the development of OER catalysts based on earth-abundant metals. Reference Chen, Wu, Han, Lin, Sun, Zhang and Cao13–Reference Du, Chen, Ye, Wiley and Meyer15
Over the past decade, significant advances have been achieved in developing water oxidation catalysts based on the first–row transition metals such as manganese, Reference Tagore, Crabtree and Brudvig16 iron, Reference Chen, Wu, Han, Lin, Sun, Zhang and Cao13,Reference Ellis, McDaniel, Bernhard and Collins17 cobalt, Reference McAlpin, Surendranath, Dinca, Stich, Stoian, Casey, Nocera and Britt18 and nickel, Reference Joya, Joya and de Groot14,Reference Dinca, Surendranath and Nocera19 due to their high abundance and low cost. Besides these earth-abundant elements, copper is an attractive low-cost metal and is relatively less harmful to the environment compared with elements like cobalt and nickel. However, it is only recently that a handful of Cu-based electrocatalysts have been shown to electrocatalyze water oxidation in weakly basic to basic solutions. Reference Du, Chen, Ye, Wiley and Meyer15,Reference Chen, Gao, Lu, Ye and Sun20–Reference Zhang, Chen, Kang and Meyer27
To obtain more economical and efficient catalysts for practical applications, the catalysts should be prepared in a way that is as simple as possible. Reference Liu, Cui, Sun and Du23,Reference Zhang, Qi, Liu and Cao28–Reference McKone, Sadtler, Werlang, Lewis and Gray30 Recently, some methods have been developed to prepare electrocatalyst electrodes for water oxidation, such as hydrothermal or solid–state reactions Reference Cui, Liu, Sun and Du22,Reference Qi, Zhang, Xiang, Liu, Wang, Chen, Han and Cao31 and electrodeposition. Reference Liu, Jia, Sun, Chen, Xu and Du24–Reference Zhang, Chen, Kang and Meyer27,Reference Liu, Sun, Cui and Du32 The Cu-based electrocatalysts prepared by hydrothermal or solid–state reactions are typically powders, which need the aid of polymeric binders, for example, Nafion, to bind them to the electrode substrate. Reference Cui, Liu, Sun and Du22 However, the utilization of polymeric binders can occupy active sites of the catalyst and deteriorate the electrical conductivity of the electrode, leading to a reduced electrocatalytic performance. Reference Lu and Zhao33,Reference Wang, Ji and Chen34 The electrodeposition method has the advantage of integrating the synthesis of the catalyst and subsequent film formation into one facile electrochemical step without the need for binding agents. Reference Chen, Gao, Lu, Ye and Sun20,Reference Liu, Jia, Sun, Chen, Xu and Du24,Reference Liu, Sun, Cui and Du32 However, the dissolution of the Cu(II) ion [K sp(Cu(OH)2) = 2.2 × 10−20 or K sp(Cu3(PO4)2) = 1.40 × 10−37] in alkaline or neutral buffer solutions limits the use of simple Cu(II) ions as precursors to prepare the OER electrode by electrodeposition. Reference Du, Chen, Ye, Wiley and Meyer15 Consequently, researchers have to bypass the obstacle by using the pre-prepared, soluble copper complexes as precursors for electrodeposition. By contrast, the electrophoresis method could directly use metal hydroxide colloids for the electrode preparation, which are usually the active species toward OER, and requires neither a laborious catalyst precursor synthesis nor the aid of polymeric binders. To the best of our knowledge, there have been no reports through the facile electrophoresis deposition (EPD) method for the fabrication of Cu-based electrocatalyst electrodes for OER.
Herein, we report that the Cu(OH)2-FTO electrode could be conveniently fabricated by a simple method of electrophoresis. The Cu(OH)2 nanobelts of 250 nm in diameter were synthesized by a simple chemical precipitation process. The as-prepared Cu(OH)2 nanobelts were then immobilized onto the FTO substrate by electrophoresis in an acetone solution at room temperature. The corresponding CuO film could be obtained by calcining the Cu(OH)2 film at 300 °C in air for 2 h. The morphologies and compositions of Cu(OH)2 and CuO electrodes were characterized by a series of techniques. The electrocatalytic activity of the as-prepared Cu(OH)2-FTO and CuO-FTO electrodes was investigated in a 0.2 M phosphate buffer solution (PBS) at pH 12. The annealed CuO-FTO electrode exhibits an excellent performance for OER with an impressive onset overpotential of 370 mV and an overpotential of only 500 mV to reach a current density of 1 mA/cm2 with a low Tafel slope of 57 mV/dec. The fabrication of the surface-bound copper hydroxide and oxide catalysts via electrophoresis is facile and universal, which requires neither expensive instrumentation nor laborious catalyst precursor synthesis.
II. EXPERIMENTAL
A. Chemicals
Copper(II) chloride (CuCl2, 99.99%), sodium hydroxide (NaOH, ≥98.5%), methanol (CH3OH, 99.8%), and acetone (CH3COCH3, ≥99.5%) were purchased from Sigma Aldrich (St. Louis, Missouri). Fluorine-doped tin oxide (FTO) glass (11 Ω/sq) was obtained from Delta Technologies, Limited. All other reagents were of analytical grade and were used as received without any further purification. All electrolyte solutions were prepared with deionized water (18 MΩ cm) unless stated otherwise. The pH of PBS was adjusted with concentrated NaOH or phosphoric acid.
B. Apparatus
Powder X-ray diffraction (XRD) was measured by Bruker Foucs D8 via a ceramic monochromatized Cu Kα radiation of 1.54178 Å, operating at 40 kV and 40 mA. The scanning rate was 5° per min in 2θ, and the scanning range was from 10 to 70°. Raman spectra were recorded on a confocal microscope laser Raman spectrometer (inVia™, Renishaw, Wotton-under-Edge, United Kingdom).
Scanning electron microscope (SEM) images were obtained at Hitachi S-4800 (Hitachi, Tokyo, Japan). Transmission electron microscopy (TEM) images, high-resolution TEM (HRTEM) images, and selected area electron diffraction (SAED) patterns were obtained using JEM-2100 (JEOL Ltd., Tokyo, Japan). X-ray photoelectron spectroscopy (XPS) for elemental analysis was conducted on a Kratos Axis Ultra DLD X-ray Photoelectron Spectrometer (SHIMADZU, Kyoto, Japan). The carbon 1s peak (284.6 eV) was used for internal calibration.
Electrochemical measurements were performed on a CHI 660E electrochemical workstation (Chenhua Corp., Shanghai, China). The three-electrode system consisted of a working electrode, a platinum plate counter electrode, and a saturated calomel reference electrode (SCE, 0.244 V versus NHE). Unless stated otherwise, all potentials in cyclic voltammetry and controlled potential electrolysis (CPE) were reported versus NHE without iR compensation.
To construct a Tafel plot, the current–potential data were obtained at an FTO with an active film by linear sweep voltammetry at a very slow scan rate (0.1 mV/s) in 0.2 M PBS at pH 12. The solution resistance measured prior to the data collection (using iR test function) was used to correct the Tafel plot for the iR drop.
All experiments were performed at 20 ± 2 °C.
C. Procedures
The Cu(OH)2 colloids for electrophoresis were freshly prepared before use. Briefly, the concentrated NaOH solution was added drop by drop into the CuCl2 solution under a magnetic stirring condition at room temperature. Upon addition of NaOH, the Cu(OH)2 colloids of blue color precipitated out immediately. The Cu(OH)2 colloids were collected by centrifugation, washed with deionized water and acetone for three times, respectively, and then dried in air. Prior to electrochemical measurements, the FTO electrode was cleaned by sonication in pure water (15 min), methanol (15 min), and acetone (15 min), respectively.
D. Preparation of the Cu(OH)2-FTO and CuO-FTO electrodes
EPD method was utilized to prepare Cu(OH)2 films on FTO. During EPD, the cleaned FTO glass was used as the working electrode by keeping it at a positive potential (anode), while a pure steel mesh was used as the counter (cathode) electrode. The linear distance between the two electrodes was around 1 cm. The electric power was supplied by a programmable DC-regulated power-stabilized current supply (DH1765-3). The applied voltage was 10 V and the time for EPD varied. The electrolyte solution consists of 4 mg/L of I2, 16 mg/L of Cu(OH)2, and 20 mL acetone, and the temperature of the electrolyte solution was kept constant at 25 °C. After EPD, the as-prepared Cu(OH)2-FTO electrode was dried in air. The as-prepared Cu(OH)2-FTO electrodes by EPD were heated at 300 °C in air for 2 h to obtain the CuO-FTO electrodes.
III. RESULTS AND DISCUSSION
A. Characterization of the catalyst films
The blue Cu(OH)2 precipitates were firstly prepared by a simple aqueous solution reaction of copper chloride and sodium hydroxide. In general, thin films with small amounts of the catalyst make it hard for the material to reach its full catalytic potential, while films that are too thick may block the electron transfer to the underlying substrate and weaken the catalytic performance. So, it is crucial to optimize the thickness of the catalyst film for optimum catalytic performance with an optimized electrodeposition time. As shown in Fig. 1(a), the Cu(OH)2-FTO electrode by electrophoresis is in blue color, and the thickness of the Cu(OH)2 film gradually increases with increasing the electrophoresis time. Correspondingly, the CuO-FTO electrode was prepared by calcination of the as-obtained blue Cu(OH)2 film. As shown in Fig. 1(b), the blue Cu(OH)2 films were transformed to the dark orange CuO films due to the dehydration.

FIG. 1. (a) Camera photos of the Cu(OH)2-FTO electrodes prepared by electrophoresis for different times. (b) Corresponding to (a), the CuO-FTO electrodes by further calcination at 300 °C for 2 h.
The morphologies of the Cu(OH)2-FTO and CuO-FTO electrodes were examined by SEM. In Fig. 2(a), the as-synthesized Cu(OH)2 materials feature uniform nanobelt structures with an average diameter of ∼250 nm. In Fig. 2(b), the CuO materials by calcination display a nanowire-like structure with an average diameter of ∼50 nm. In addition, the diameter of CuO is smaller than that of Cu(OH)2, and the overall structure of CuO is more fluffy and irregular. The loose nanowire-like structure of CuO probably leads to a relatively high specific surface area, which may greatly increase the total surface area and enhance the catalytic current density (see below). The cross-sectional profile of the Cu(OH)2-FTO and CuO-FTO electrodes were also examined by SEM. As shown in Figs. 2(c) and 2(d), the average thickness of the catalytic films is approximately 1 μm, and the structure of the Cu(OH)2 film is consistently more fluffy than that of the CuO film.

FIG. 2. Top-down SEM images of (a) Cu(OH)2-FTO and (b) CuO-FTO electrodes. Cross-sectional SEM images of (c) Cu(OH)2-FTO and (d) CuO-FTO electrodes.
The morphological structure of Cu(OH)2 and CuO were further demonstrated by TEM and HRTEM images. In Fig. 3(a), the typical TEM image shows that the nanobelt structure of Cu(OH)2 is built up by the accretion of straight nanowires with uniform diameters of 50 nm. The SAED pattern of the Cu(OH)2 material shows apparent diffraction rings [inset of Fig. 3(a)], indicating the polycrystalline nature of the material. The HRTEM image in Fig. 3(b) demonstrates a fringe spacing of 0.24 nm in certain domains, corresponding to the (002) plane of Cu(OH)2. Reference Wen, Zhang and Yang35

FIG. 3. TEM and HRTEM images of (a, b) Cu(OH)2 and (c, d) CuO. The insets in (a) and (c) are the corresponding SAED patterns.
The TEM image of the CuO material is also shown in Fig. 3(c). Consistent with the SEM image, the sample of CuO displays somewhat a fluffy structure, and these nanowires consist of interconnected nanoparticles. The SAED pattern of these nanowires show discernible diffraction rings and spots [inset of Fig. 3(c)], indicating that the nanowires are of polycrystalline structure. The HRTEM image in Fig. 3(d) demonstrates a fringe spacing of 0.264 nm in certain domains, which agrees well with the spacing of the
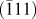 $\left( {\bar{1}11} \right)$
lattice plane of CuO.
Reference Wang, Varghese, Ruan, Paulose and Grimes36
$\left( {\bar{1}11} \right)$
lattice plane of CuO.
Reference Wang, Varghese, Ruan, Paulose and Grimes36
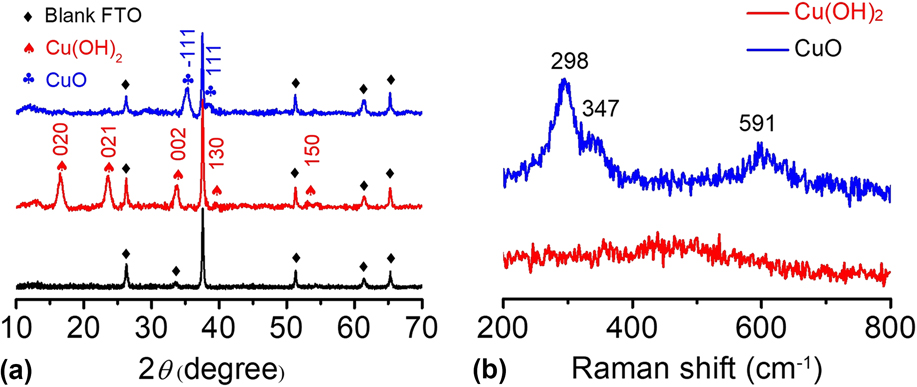
The Cu(OH)2 and CuO materials were further characterized by other analysis techniques. In Fig. 4(a), the XRD pattern shows that the main peaks of Cu(OH)2 are located at 16.5°, 23.6°, 26.3°, 33.8°, 37.5°, and 51.3° in 2θ, corresponding to the (020), (021), (002), (111), (130), and (150) planes, respectively.
Reference Cui, Liu, Sun and Du22
The diffraction peaks of the Cu(OH)2 sample were sharp, and no impurity peaks were observed except for the blank FTO background, indicating the high crystallinity and high purity of the as-prepared Cu(OH)2. After calcination, the CuO-FTO electrode displays a different XRD pattern. The main peaks are located at 35.3° and 38.6° in 2θ, corresponding to the
 $\left( {\bar{1}11} \right)$
and (111) planes, respectively.
Reference Wang, Varghese, Ruan, Paulose and Grimes36,Reference Lu, Qi, Yang, Zhang, Wu and Ma37
Apparently, the signals of Cu(OH)2 completely disappeared after calcination, indicating that Cu(OH)2 was successfully transformed into CuO. The as-prepared samples were also characterized by Raman spectroscopy. Figure 4(b) shows the Raman spectra of the Cu(OH)2-FTO and CuO-FTO electrodes. The Cu(OH)2-FTO film showed a weak and a broad characteristic peak at 490 cm−1, and the CuO-FTO films exhibited three peaks at 298, 347, and 591 cm−1, respectively. Similar Raman spectra of Cu(OH)2 and CuO materials have also been recorded in a previous report.
Reference Deng, Handoko, Du, Xi and Yeo38
$\left( {\bar{1}11} \right)$
and (111) planes, respectively.
Reference Wang, Varghese, Ruan, Paulose and Grimes36,Reference Lu, Qi, Yang, Zhang, Wu and Ma37
Apparently, the signals of Cu(OH)2 completely disappeared after calcination, indicating that Cu(OH)2 was successfully transformed into CuO. The as-prepared samples were also characterized by Raman spectroscopy. Figure 4(b) shows the Raman spectra of the Cu(OH)2-FTO and CuO-FTO electrodes. The Cu(OH)2-FTO film showed a weak and a broad characteristic peak at 490 cm−1, and the CuO-FTO films exhibited three peaks at 298, 347, and 591 cm−1, respectively. Similar Raman spectra of Cu(OH)2 and CuO materials have also been recorded in a previous report.
Reference Deng, Handoko, Du, Xi and Yeo38
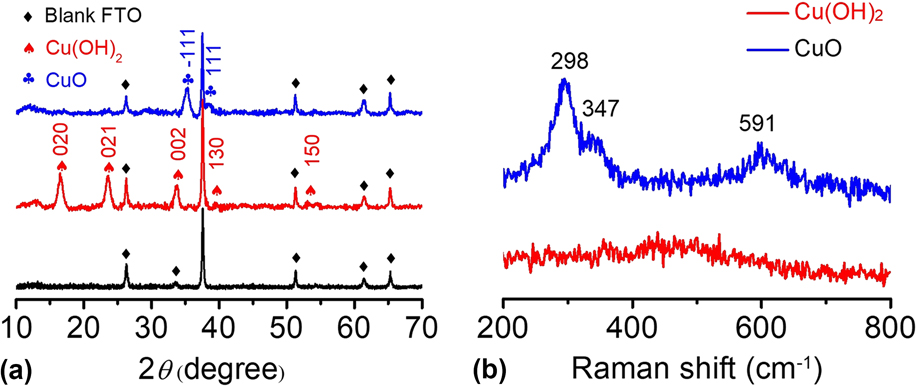
FIG. 4. (a) XRD patterns of blank FTO, Cu(OH)2-FTO and CuO-FTO electrodes. (b) Raman spectra of the Cu(OH)2-FTO and CuO-FTO electrodes.
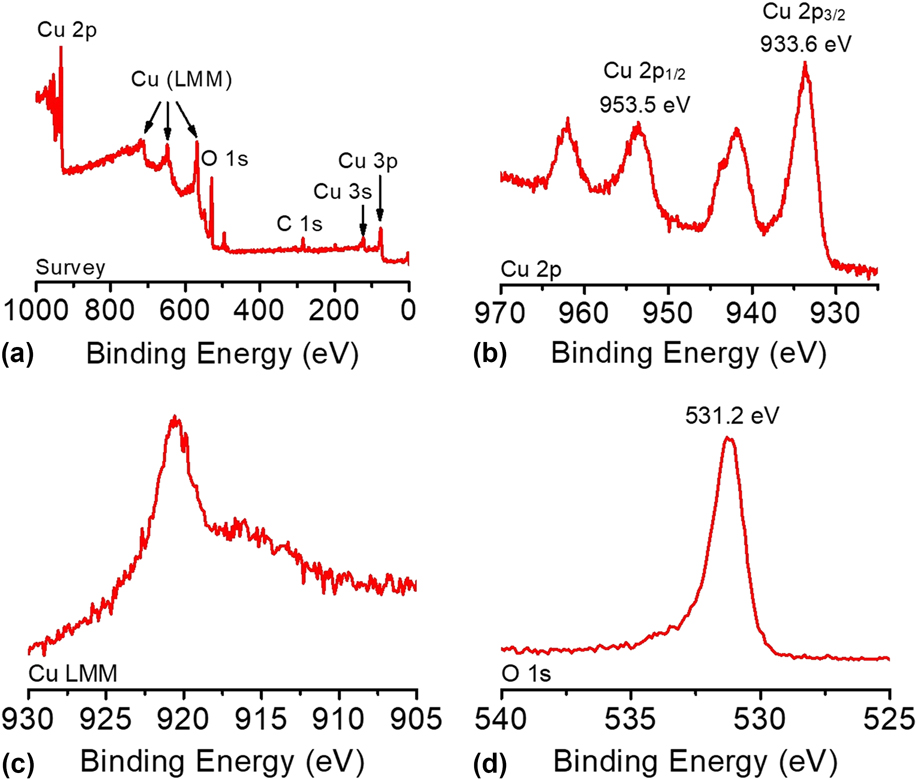
The composition and oxidation state of the CuO-FTO electrode were elucidated by XPS measurements. The survey XPS spectrum in Fig. 5(a) corroborates the coexistence of Cu and O on the electrode surface with an atomic ratio of ∼1:1 for Cu and O, which is consistent with the presence of CuO. Figures 5(b)–5(d) shows the high-resolution XPS spectra of Cu 2p, Cu LMM, and O 1s, respectively. In Fig. 5(b), the Cu 2p XPS spectrum displayed two dominant peaks at 933.6 and 953.5 eV, accompanied by two less intense satellite peaks, consistent with the presence of divalent Cu rather than Cu(0). Reference Yu, Li, Zhang, Li and Sun26,Reference Lu, Du, Su, Zhang, Xu, Meyer and Chen39,Reference Durando, Morrish and Muscat40 The two determined binding energies are assigned to Cu 2p 1/2 and Cu 2p 3/2, which are consistent with CuO. In this study, Cu(OH)2 was ruled out, which has been reported to exhibit 2p 1/2 and 2p 3/2 peaks at more positive binding energies by 1.2–1.5 eV. Reference Yu, Li, Zhang, Li and Sun26,Reference McIntyre, Sunder, Shoesmith and Stanchell41 The presence of CuO can be further confirmed by the presence of a peak at 916.8 eV in the Cu LMM spectrum and a well-resolved O 1s peak observed at 531.2 eV, as shown in Figs. 5(c) and 5(d). Reference Cui, Liu, Sun and Du22,Reference Joya and de Groot42
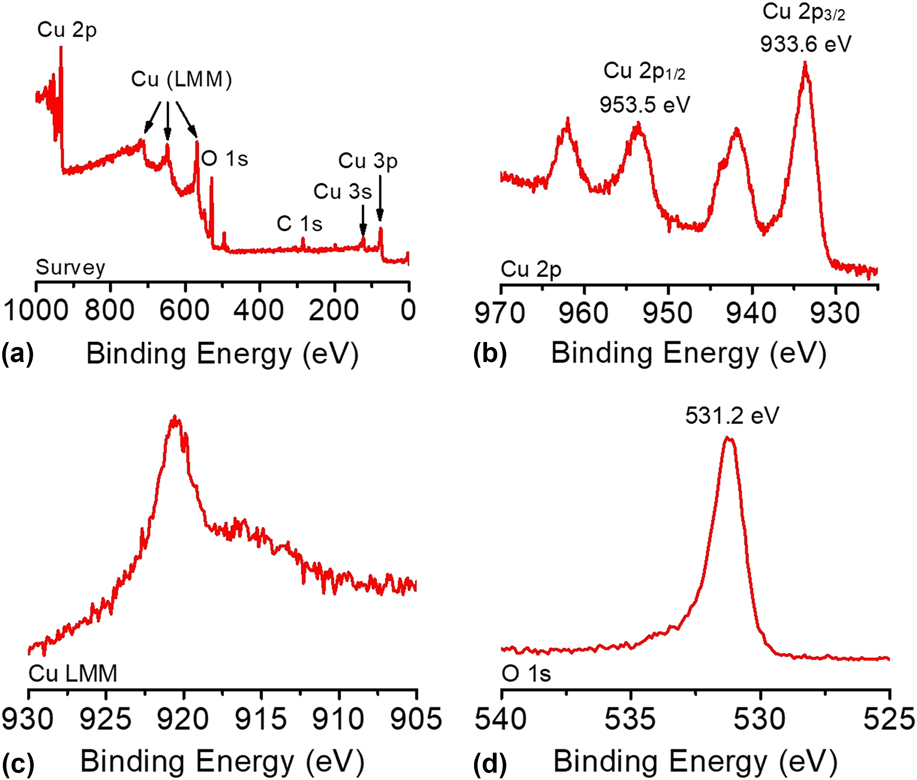
FIG. 5. (a) The survey XPS spectrum and the high-resolution XPS spectra of (b) Cu 2p, (c) Cu LMM, and (d) O 1s of the CuO-FTO electrode.
B. Electrocatalytic water oxidation
The catalytic performance of the electrodes was investigated using a standard three-electrode system in 0.2 M PBS at pH 12. To determine the optimal electrophoresis time, the cyclic voltammograms (CVs) of the bare FTO and the Cu(OH)2-FTO electrodes prepared by different electrophoresis times were recorded in Fig. 6(a). The blank FTO shows a negligible current density within the studied potential range. By contrast, the Cu(OH)2-FTO electrodes exhibit significantly enhanced catalytic currents dependent on the electrophoresis time. In general, the catalytic activity of the electrode increased rapidly when the electrophoresis time was initially increased, and it then gradually decreased by further prolonging the electrophoresis time. A continuously increasing film thickness by prolonging the electrophoresis time can be easily affirmed in Fig. 1(a). The thick film may block electron transfer to the underlying substrate, resulting in the decrease of catalytic current. With an optimized electrophoresis time of 10 s, the catalytic current density is maximized, as shown in Fig. 6(a) and its inset. A current density of 4.3 mA/cm2 is achieved at a potential of 1.3 V versus NHE.
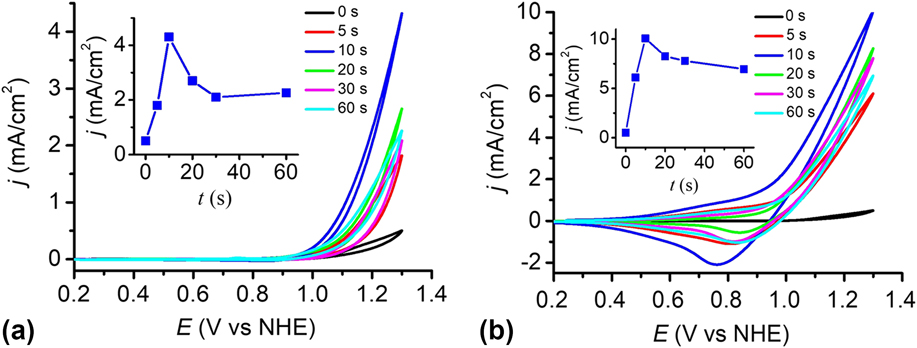
FIG. 6. (a) CVs at the Cu(OH)2-FTO electrodes prepared by electrophoresis for different time in 0.2 M PBS at pH 12. (b) As in (a), CVs at the CuO-FTO electrodes. Scan rate: 100 mV/s. The insets in (a) and (b) show the corresponding plots of catalytic current densities of the Cu(OH)2-FTO and CuO-FTO electrodes at 1.3 V versus the electrophoresis time.
The catalytic performance of the CuO-FTO electrodes prepared by calcination of the Cu(OH)2-FTO electrodes at 300 °C for 2 h was also investigated in 0.2 M PBS at pH 12. As shown in Fig. 6(b) and its inset, the CuO-FTO electrode prepared by 10 s electrophoresis also shows the excellent electrocatalytic activity for OER. A current density of 10 mA/cm2 was obtained at a potential of 1.3 V versus NHE. The performance of the CuO-FTO electrode substantially outperformed the Cu(OH)2-FTO electrode under the similar experimental condition. The higher catalytic current density obtained at CuO-FTO than at Cu(OH)2-FTO may be at least partially attributed to the rough nanowire structure of CuO that exposes more catalytic active sites than the smooth nanobelt-like Cu(OH)2. Interestingly, as shown in the reverse/cathodic scan of CV, a clear cathodic peak can be observed at ∼0.80 V, which is probably due to the reduction of a high-oxidation-state Cu intermediate, which is responsible for the water oxidation catalysis. Reference Cui, Liu, Sun and Du22,Reference Liu, Jia, Sun, Chen, Xu and Du24,Reference Zhang, Chen, Kang and Meyer27
To test the stability of the Cu(OH)2-FTO and CuO-FTO electrodes for OER, CPE experiments were conducted at 1.30 V versus NHE in 0.2 M PBS at pH 12. The efficiency of electrocatalytic water oxidation at the electrode surface is sufficient such that O2 gas bubbles were observed once the potential was applied. As shown in Fig. 7(a), the Cu(OH)2-FTO electrode displayed a current density of 0.8 mA/cm2 at a potential of 1.2 V. Interestingly, a sustained current density of 4 mA/cm2 was achieved at the CuO-FTO electrode under the same condition, which is consistent with the results of the CVs test. The constant currents obtained at both Cu(OH)2-FTO and CuO-FTO electrodes indicated that the morphological structure and catalytic activity of the materials were well retained under the water oxidation conditions.
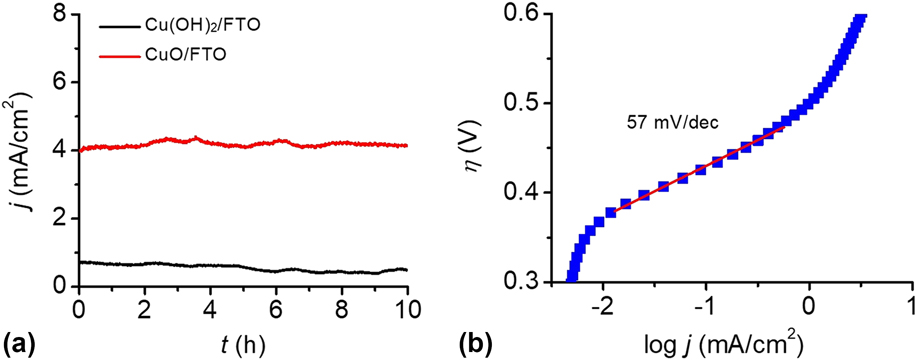
FIG. 7. (a) CPEs of the Cu(OH)2-FTO and CuO-FTO electrodes at 1.2 V in 0.2 M PBS at pH 12. (b) The Tafel plot of an as-prepared CuO-FTO electrode in 0.2 M PBS at pH 12, corrected for the iR drop of the solution.
Tafel measurements were conducted to evaluate the intrinsic catalytic activity of the CuO-FTO electrode toward OER. The Tafel slope was obtained from the polarization curve (0.1 mV/s) using a linear fit applied to points in the Tafel region. In the curve, the overpotential was determined by the equation η = V appl − iR − E pH, where V appl is the applied potential, i is the steady-state current, R is the uncompensated resistance, and E pH is the thermodynamic potential for water oxidation at this pH (E pH = 1.23 V − 0.059 pH V versus NHE). As shown in Fig. 7(b), the CuO-FTO electrode displays a Tafel slope of 57 mV/dec in the 0.2 M PBS solution. From the Tafel plot, an appreciable catalytic current is observed starting at η = 370 mV, and a current density of 1 mA/cm2 required η = 500 mV. The low overpotential and Tafel slope make this material among the most active Cu-based heterogeneous electrocatalysts in weakly basic to basic solutions, note Table I.
TABLE I. Comparison of the heterogeneous Cu-based electrocatalysts for water oxidation.

TEOA = triethanolamine; en = 1,2-ethylenediamine; Gly = Glycine.
C. pH effect
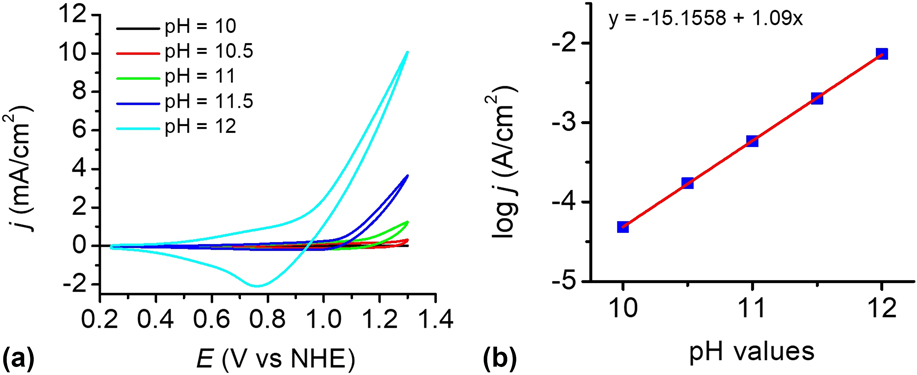
To investigate the influence of pH, the catalytic reactivity of the CuO film was measured in 0.2 M phosphate buffer solutions at different pH values. Figure 8(a) shows the dependence of the current density on pH values in the range of pH 8–12. The electrocatalyst film exhibits an excellent catalytic property in PBS at pH 12 than others. Within the pH range of 10–12, a linear relationship between log j and pH was observed at 1.2 V versus NHE, giving a slope of 1.09 decade/pH, as shown in Fig. 8(b). According to the Tafel slope and the slope of the log j—pH plot, the pH effect on the catalytic potential of the CuO film was calculated by applying Eq. (1). Reference Yu, Li, Zhang, Li and Sun26 The variation of the catalytic potential with pH was deduced to be −62 mV/pH, which is close to the value of 59 mV/pH. This value agrees well with the Nernstian behavior, suggesting a possible 2e−/2H+ transfer prior to a chemical rate-determing step. Reference Chen, Gao, Lu, Ye and Sun20,Reference Yu, Li, Zhang, Li and Sun26,Reference Tae, Song, Lee, Kim, Yoon, Hwang, Kim and Yoon44–Reference Zhang, Wu, Qi, Chen and Cao47
 $${\left( {{{\partial E} \over {\partial {\rm{pH}}}}} \right)_j} = - {\left( {{{\partial \log j} \over {\partial {\rm{pH}}}}} \right)_E}{\left( {{{\partial E} \over {\partial \log j}}} \right)_{{\rm{pH}}}}\quad .$$
$${\left( {{{\partial E} \over {\partial {\rm{pH}}}}} \right)_j} = - {\left( {{{\partial \log j} \over {\partial {\rm{pH}}}}} \right)_E}{\left( {{{\partial E} \over {\partial \log j}}} \right)_{{\rm{pH}}}}\quad .$$
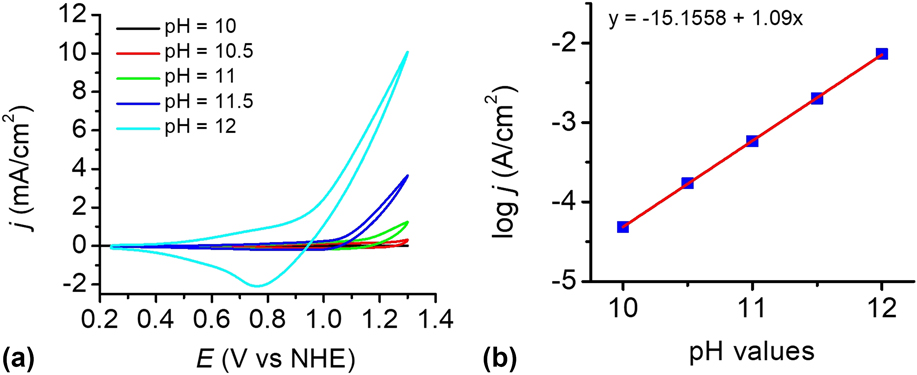
FIG. 8. (a) CVs of the as-prepared CuO-FTO electrode in 0.2 M PBS at different pH values. Scan rate: 100 mV/s. (b) Current density dependence on pH value at 1.2 V versus NHE in 0.2 M PBS without iR compensation.
IV. CONCLUSIONS
In summary, we have used a simple aqueous solution reaction and electrophoresis process to fabricate Cu-based electrodes as efficient electrocatalysts for water oxidation. Through comparison of Cu(OH)2 and CuO catalyst films, we found that the CuO film exhibits a superior catalytic activity for water oxidation. At the CuO-FTO electrode, a current density of 4 mA/cm2 was sustained for at least 10 h in 0.2 M PBS. The morphology and structure of the film electrodes were also investigated through a series of techniques. In addition, through investigation of the pH effect, the catalytic potential-pH dependence was determined to be −62 mV/pH, which is close to the expected value of 59 mV/pH. The facile electrocatalyst preparation strategy developed in this study is important for the real application of the Cu-based catalyst film, and this strategy might also be extended to prepare other metal oxide electrocatalysts.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (21573160 and 21405114) and Science & Technology Commission of Shanghai Municipality (14DZ2261100).