



The Alphafold2 method (Jumper et al., Reference Jumper, Evans, Pritzel, Green, Figurnov, Ronneberger, Tunyasuvunakool, Bates, Žídek, Potapenko, Bridgland, Meyer, Kohl, Ballard, Cowie, Romera-Paredes, Nikolov, Jain, Adler, Back, Petersen, Reiman, Clancy, Zielinski, Steinegger, Pacholska, Berghammer, Bodenstein, Silver, Vinyals, Senior, Kavukcuoglu, Kohli and Hassabis2021a) has unquestionably revolutionized the field of protein structure prediction, achieving very high accuracy for most targets during the CASP14 initiative (Jumper et al., Reference Jumper, Evans, Pritzel, Green, Figurnov, Ronneberger, Tunyasuvunakool, Bates, Žídek, Potapenko, Bridgland, Meyer, Kohl, Ballard, Cowie, Romera-Paredes, Nikolov, Jain, Adler, Back, Petersen, Reiman, Clancy, Zielinski, Steinegger, Pacholska, Berghammer, Silver, Vinyals, Senior, Kavukcuoglu, Kohli and Hassabis2021b). Recently, the Alphafold2 strategy has been extended to predict protein–protein complexes (Evans et al., Reference Evans, O'Neill, Pritzel, Antropova, Senior, Green, Žídek, Bates, Blackwell, Yim, Ronneberger, Bodenstein, Zielinski, Bridgland, Potapenko, Cowie, Tunyasuvunakool, Jain, Clancy, Kohli, Jumper and Hassabis2021). In the article presenting the multimer version of Alphafold2, a set of 17 complexes obtained after the network training date was considered to compare with previous strategies based on the initial Alphafold2 system (Ghani et al., Reference Ghani, Desta, Jindal, Khan, Jones, Kotelnikov, Padhorny, Vajda and Kozakov2021). Out of these 17 complexes, Alphafold2 multimer achieved correct predictions for 14 cases, as assessed by the DockQ score (Basu and Wallner, Reference Basu and Wallner2016).
I looked at the three cases where Alphafold2 produced models with low DockQ scores (Evans et al., Reference Evans, O'Neill, Pritzel, Antropova, Senior, Green, Žídek, Bates, Blackwell, Yim, Ronneberger, Bodenstein, Zielinski, Bridgland, Potapenko, Cowie, Tunyasuvunakool, Jain, Clancy, Kohli, Jumper and Hassabis2021). In the first version of the study (Evans et al., Reference Evans, O'Neill, Pritzel, Antropova, Senior, Green, Žídek, Bates, Blackwell, Yim, Ronneberger, Bodenstein, Zielinski, Bridgland, Potapenko, Cowie, Tunyasuvunakool, Jain, Clancy, Kohli, Jumper and Hassabis2021), the three failed cases were 5ZNG (DockQ = 0.02), 6A6I (DockQ = 0.05), and 7NLJ (DockQ = 0.06).Footnote 1 I reproduced Alphafold2 predictions with a local installation of the ParaFold pipeline (Zhong et al., Reference Zhong, Su, Wen, Zuo, Hong and Lin2022), installed in November 2021, excluding the templates newer than April 2018 (--max_template_date = 2018-04-30, which is the network training date). Each prediction run generates five models and I considered the model with the highest confidence value as the final prediction. Overall, I am able to replicate published results, as shown in Table S1.
For 5ZNG, the predicted models are indeed very distant from the complex annotated as biological assembly in the PDB (DockQ score = 0.02). However, the five models are almost identical, and have good confidence value (around 0.7). They are very similar to the complex annotated as asymmetric unit (DockQ score = 0.7), see Fig. 1. The complex reported in the article accompanying the 5ZNG structure is indeed the one annotated as asymmetric unit in the PDB entry (Guo et al., Reference Guo, Cesari, de Guillen, Chalvon, Mammri, Ma, Meusnier, Bonnot, Padilla, Peng, Liu and Kroj2018), not the one annotated as biological assembly and generated by PISA (Krissinel and Henrick, Reference Krissinel and Henrick2007). Thus, also in this case, the Alphafold2 prediction was indeed accurate: the predicted model was indeed the one described as biologically relevant.
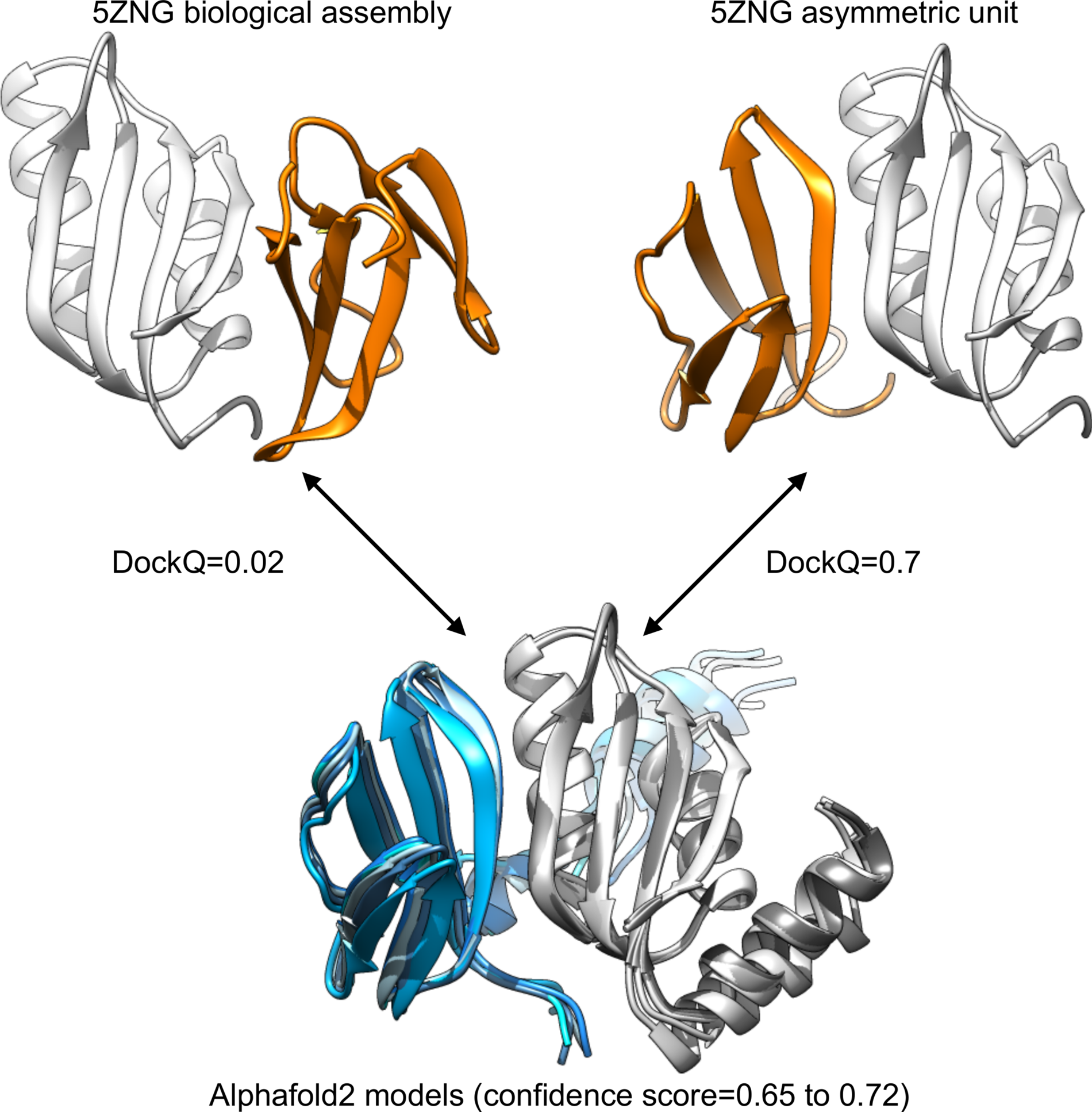
Fig. 1. Comparison between the structural information available for 5ZNG and the Alphafold2 models. The biological assembly discussed in the article introducing the structure is the one annotated as asymmetric unit in the PDB (top right).
For 6A6I and 7NLJ, I performed triplicate runs of Alphafold2 and I obtained models with highest DockQ scores than previously reported (Evans et al., Reference Evans, O'Neill, Pritzel, Antropova, Senior, Green, Žídek, Bates, Blackwell, Yim, Ronneberger, Bodenstein, Zielinski, Bridgland, Potapenko, Cowie, Tunyasuvunakool, Jain, Clancy, Kohli, Jumper and Hassabis2021): DockQ = 0.39/0.24/0.18 for 6A6I and DockQ = 0.21/0.16/0.16 for 7NLJ, with low confidence values (0.2–0.3). In these cases, the asymmetric units and biological assemblies of the PDB entries are identical, and I found no obvious reason for such discrepancy.
It is worth noting that, even if these models are far from the high quality threshold (DockQ > 0.8), they indeed provide an approximate prediction of the true binding site regions, as shown in Fig. 2.
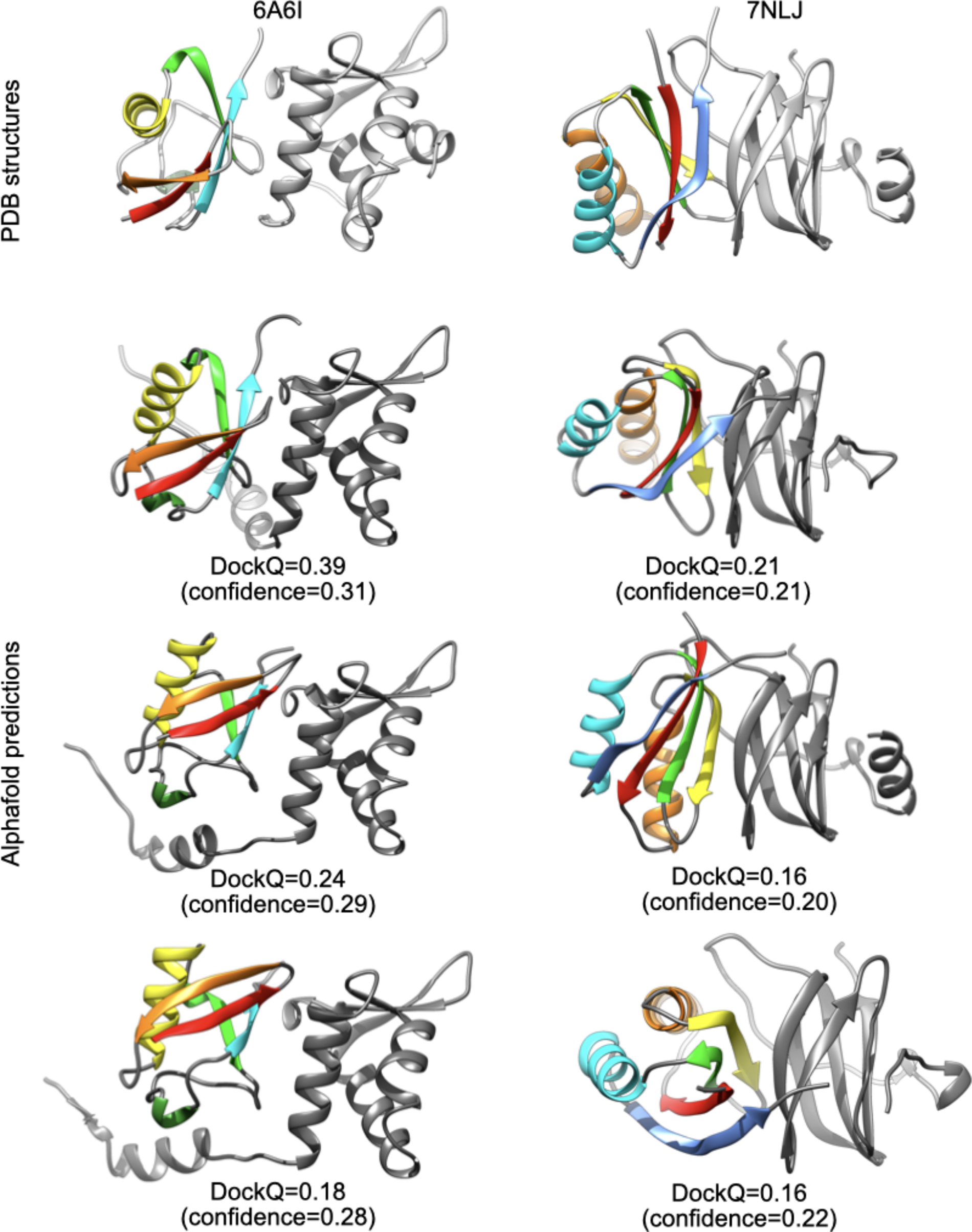
Fig. 2. Comparison between PDB structures and Alphafold2 predictions for 6A6I and 7NLJ.
In conclusion, even in the cases where Alphafold2 did not achieve correct predictions for multimers, the examination of failed cases in this very small data set suggests that the predictions could detect errors in PDB annotation (like 5ZNG) or, more interestingly, determine approximate binding sites (like 6A6I and 7NLJ). In the last case, the models could provide a good starting point for conventional docking tools with restraints to these binding sites. In addition, since Alphafold2 achieves the very difficult task of predicting both the subunit folds and their binding mode, one could wonder what accuracy it could attain in a classical docking context when the monomer structures are known. This indicates that Alphafold2 could revolutionize the field of protein–protein docking as it has done for protein structure prediction.
Acknowledgements
I gratefully acknowledge support from Alexis Michon from IBCP and the CNRS/IN2P3 Computing Center (Lyon – France) for providing computing and data-processing resources needed for this work. I gratefully acknowledge Elisa Frezza, Guillaume Launay, and Riccardo Pellarin for their constructive comments.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflict of interest
None.


