


INTRODUCTION
Human African trypanosomiasis (HAT) is endemic in sub-Saharan Africa, where millions are at risk from this disease. The disease, caused by the protozoan parasites Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense, has two stages; stage 1 is the peripheral infection, which has non-specific symptoms and stage 2 occurs when the parasite enters the central nervous system (CNS). HAT is fatal unless treated. The current treatments for stage 1 infection are suramin and pentamidine. For stage 2, the therapeutic options are melarsoprol, eflornithine and the recent combination therapy NECT (nifurtimox and eflornithine) (Brun et al. Reference Brun, Don, Jacobs, Wang and Barrett2011). The current drugs available for treating HAT are inadequate, due to toxicity and poor efficacy. There are also problems with treatment failures for melarsoprol, although it is not known if this is due to resistance (Wilkinson and Kelly, Reference Wilkinson and Kelly2009). In addition, the current treatments are inappropriate for a rural setting with poor facilities as they all require parenteral administration. There is need for new treatments for HAT, for the reasons given above, but also with the aim of elimination and eradication of this disease.
Recently there has been a marked decrease in numbers of cases and the agenda has turned more to elimination and eradication. Currently there are about 10 000 recorded cases per year. However, the true number of people afflicted by this disease is estimated to be much higher due to under-reporting; probably 30 000 cases per year (Brun et al. Reference Brun, Don, Jacobs, Wang and Barrett2011). Furthermore, it is appropriate to take warnings from history, where there has been a huge change in the number of cases; during the middle of the last century the disease was nearly eliminated, but was resurgent, and, by the end of the last century, there were an estimated 450 000 people affected (Barrett, Reference Barrett1999, Reference Barrett2006; Brun et al. Reference Brun, Don, Jacobs, Wang and Barrett2011).
There have been very few drug discovery and development programmes for HAT. Recently, however, there has been an increased effort in drug discovery for HAT and other neglected tropical diseases due to the generosity of organizations such as the Wellcome Trust, the Bill and Melinda Gates Foundation and the organization Drugs for Neglected Diseases (DNDi). This has led to increased understanding of the drug discovery processes for diseases such as HAT and infrastructure put into place for drug discovery. Given this impetus, it is now important to make sure that new medicines are delivered for HAT.
Significant recent approaches include a diamidine developed by Tidwell's group, which was taken as far as phase 3 clinical trials, but unfortunately had to be withdrawn (Barrett and Croft, Reference Barrett and Croft2012). Two new molecules have been moved into clinical trials by DNDi. These compounds are the nitro heterocycle fexinadazole and the oxaborole SCYX-7158. This is an exciting new development.
Drug discovery for neglected tropical diseases, such as HAT, is being conducted using both target-based and phenotypic approaches (Gilbert et al. Reference Gilbert, Leroy and Frearson2011). In this article, approaches to selection of targets and molecules for target-based drug discovery are discussed. Critical to the selection of targets and molecules is the Target Product Profile.
TARGET PRODUCT PROFILE
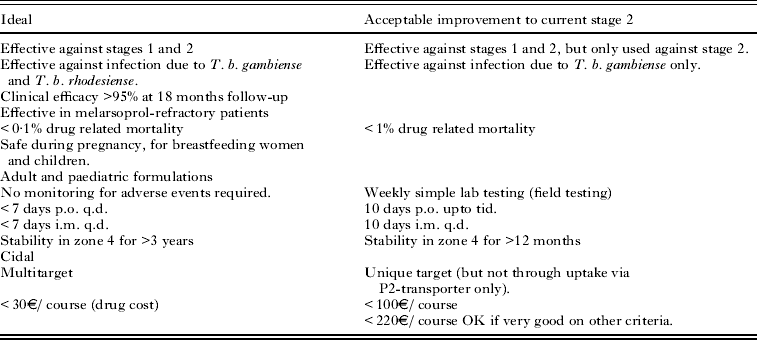
Target product profiles (TPP) define the requirements for a drug molecule to be clinically applicable. These include features such as the acceptable route for dosing the compound, the duration of treatment, the patient population, cost and acceptable range of toxicity. These are defined by clinicians and public health agencies who understand the clinical needs of the patients (Wyatt et al. Reference Wyatt, Gilbert, Read and Fairlamb2011). An example of a TPP for HAT, defined by DNDi is shown in Table 1. The TPP needs to be regularly reviewed, as it may well change due to other treatment options becoming available, changes in the clinical situation or drug resistance to existing compounds becoming a significant issue.
Table 1. Example of a new stage 1+2 target product profile (produced by DNDi)

The target product profile will inform drug discovery projects from the very start. This helps to define both the molecular target and chemical matter that is developed during a drug discovery programme.
Thus, the molecular target must be able to fulfil the TPP. For example, in the case of HAT, the molecular target must: (1) be essential for viability of the parasite in the human host; (2) have a rapid cidal effect when inhibited and (3) be amenable to inhibition by small drug-like molecules that have the correct physiochemical properties to be orally bioavailable and cross the blood-brain barrier.
Similarly, the molecules that are for screening must be capable of fulfilling the TPP once optimized through the hits to lead and lead optimization phase. In the case of HAT for example, there is a need for orally bioavailable compounds; thus compounds that are selected should have the appropriate physicochemical and pharmacokinetic properties for oral bioavailability.
Therefore in target-based drug discovery, in order to fulfil the TPP, both the molecular target and the chemical matter must be carefully selected, and their selection actually go hand-in-hand.
SELECTION OF DRUG TARGETS
The selection of a target for drug discovery is of key importance. A target is considered truly validated when it is in the clinic for treating human diseases. In the case of HAT (and the other kinetoplastid diseases), there is really only one target fully validated to this extent, ornithine decarboxylase. This is the target of the drug eflornithine (DFMO), which is used for the treatment of HAT. There are other drug targets which have significant levels of validation though; for example, protein farnesyltransferase (Eastman et al. Reference Eastman, Buckner, Yokoyama, Gelb and Van Voorhis2006; Gillespie et al. Reference Gillespie, Yokoyama, Lu, Eastman, Bollinger, Van Voorhis, Gelb and Buckner2007) and S-adenosylmethionine decarboxylase (Jacobs et al. Reference Jacobs, Nare and Phillips2011; Velez et al. Reference Velez, Brautigam and Phillips2013). Another target for HAT is N-myristoyltransferase (NMT) that we have pursued at the Drug Discovery Unit (DDU) at the University of Dundee (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, Van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010; Brand et al. Reference Brand, Cleghorn, McElroy, Robinson, Smith, Hallyburton, Harrison, Norcross, Spinks, Bayliss, Norval, Stojanovski, Torrie, Frearson, Brenk, Fairlamb, Ferguson, Read, Wyatt and Gilbert2012).
Current methods for target-based drug discovery usually involve screening diverse or focused libraries against the molecular target. The hits are then characterized and optimized through rounds of design, synthesis and testing against protein and cell. The testing usually includes assays for potency, selectivity and pharmacokinetic properties. In terms of target validation, it is important to prove that the compounds are acting on-target, and that it is inhibition (or modulation) of the target that is causing the death of the parasite. The compounds are then assayed in animal models of infection. It is only at this stage that it becomes clear if the proposed drug target has a reasonable degree of validation. In other words, a very considerable amount of time and resource will be consumed before there is reasonable evidence that the target chosen is a valid drug target. Therefore it is important to choose drug targets very carefully at the outset of a drug discovery programme.
TRAFFIC LIGHT SYSTEM
At the DDU at the University of Dundee, we have carried out target-based drug discovery programmes against HAT. We use 6 criteria to help us select molecular targets to enter our drug discovery pipeline. The criteria are: essentiality, druggability, assayability, resistance potential, toxicity potential and structural information. To help compare between the different targets, we have established a ‘traffic light’ scoring system for the different criteria, as reported (Frearson et al. Reference Frearson, Wyatt, Gilbert and Fairlamb2007; Wyatt et al. Reference Wyatt, Gilbert, Read and Fairlamb2011). Essentially each criterion is given a green, amber or red score, according to a number of definitions, which are summarized in Table 2. This provides a rapid visual method to assess an individual target and to compare between different potential drug targets. The assignment of a ‘red’ score does not necessarily mean that this is a no-go situation; it could just indicate a lack of information. There are also clear cases for a no-go decision – for example, if it is shown that the target is not essential for survival of the parasite.
Table 2. Criteria for molecular targets developed at the DDU (Frearson et al. Reference Frearson, Wyatt, Gilbert and Fairlamb2007; Wyatt et al. Reference Wyatt, Gilbert, Read and Fairlamb2011)
Essentiality
When designing a compound against a pathogen, it is important that the drug target is essential. This can be assessed both genetically and chemically. Genetic validation can be carried out in a number of ways. In many organisms this can be a complex process. In the case of T. brucei, there are more genetic tools available than with many other organisms; however, even in the case of this organism, genetic validation can still be problematic. In the case of T. brucei, it is possible to carry out RNAi to knock down the enzyme and see if there is a phenotypic response (Motyka and Englund, Reference Motyka and Englund2004; Ullu et al. Reference Ullu, Tschudi and Chakraborty2004). A more rigorous method is to carry this out by knockout studies (Barrett et al. Reference Barrett, Mottram and Coombs1999; Beverley, Reference Beverley2003).
Whilst knockdown or knockout of a gene in cell culture can prove that a gene is essential to the viability of the parasite, it does not necessarily mean that it is a good drug target. There can be issues with druggability (see below). Other reasons that the genetic validation may not be sufficient include by-pass mechanisms that can occur in vivo. Furthermore, during knockout experiments the protein is removed. The protein may have a vital ‘structural’ role due to interactions with other proteins, rather than the reaction products being vital to the parasite. A further complication with genetic validation is the rate of kill. So for example, the enzyme trypanothione synthase is genetically essential for the viability of the parasite. However, genetic knockdown/knockout of the enzyme leads to a slow rate of killing of T. brucei, meaning an inhibitory drug concentration would have to be maintained for an extended period of time before any therapeutic effect is likely to be observed (Spinks et al. Reference Spinks, Torrie, Thompson, Harrison, Frearson, Read, Fairlamb, Wyatt and Gilbert2012).
Druggability
It is important that it is possible to obtain drug-like inhibitors of the enzyme active site that have the potential to fulfil the TPP. For example, if the TPP requires an oral drug, the binding pocket of the molecular target should have a good balance of polarity and hydrophobicity, and also present a reasonably-sized pocket. The pocket should not be too large, as this will probably require a large molecule to obtain good inhibition of the target. Similarly, for a pocket that is too small it may be difficult to obtain an inhibitor with sufficient selectivity and potency. This will mean that the inhibitor (which will have complementarity in both shape and charge distribution to the active site) is more likely to have the properties required for an oral drug (see below for more discussion). The best evidence for druggability is that there is clinical precedence for this target class. Whilst this level of information is not available for most targets, often there are literature reports of inhibitors or tool molecules that inhibit the target or close homologues of the target which guide target assessment. There are also computational methods to assess druggability, such as one produced at Dundee by Krasowski et al. (Reference Krasowski, Muthas, Sarkar, Schmitt and Brenk2011). Recently it has been reported that fragment screening is an effective way to assess druggability (Edfeldt et al. Reference Edfeldt, Folmer and Breeze2011).
Assayability
Developing robust assays, suitable for 384-well format screening can be a time-consuming challenge. There is now a large array of different screening platforms available and, for most enzymes, it is possible to derive an appropriate assay. A more significant problem is actually obtaining soluble, active proteins to carry out assays. Protein expression can be very challenging and we have identified a number of targets we were interested in investigating, but for which we were unable to obtain soluble, active proteins.
Resistance potential
Resistance is always going to be a problem with anti-infectives. There are many different ways by which resistance can occur; for example, single point mutations altering residues in the target, efflux pumps, over-expression of the target, gene amplification of the target, inactivation of the drug. Some of these are very difficult to predict but, in some cases, there are clear pathways available to resistance, such as isoforms within the same organism. In the case of many organisms it is possible to check this, through bioinformatic analysis of the genomes.
Toxicity potential
Toxicity can arise from a number of different causes. Toxicity can occur if there is a human homologue of the enzyme that is being targeted. It can be difficult to predict if compounds will inhibit both the pathogen enzyme and human homologue. Occasionally even if the pathogen and human enzymes have very similar enzyme structures, it can be possible to obtain highly selective inhibitors of one enzyme, for reasons that are not well understood. For example, in the DDU compounds were obtained that are very selective for the leishmania protein kinase CRK3, although it has a similar sequence in the active site to various human protein kinases (Cleghorn et al. Reference Cleghorn, Woodland, Collie, Torrie, Norcross, Luksch, Mpamhanga, Walker, Mottram, Brenk, Frearson, Gilbert and Wyatt2011). Nevertheless, where there is a high level of similarity between the active sites of pathogen and human homologues, this should act as a significant risk factor when selecting potential drug targets in the absence of other information.
Toxicity can also arise from other unexpected interactions of compounds with the human host. Sometimes this can be deduced from the structure of the compound; hence many organizations remove potential toxic functionality (such as Michael acceptors) from their screening collections. Other toxicities are not easy to predict, and are often only picked up much later in the drug discovery process. Toxicity is one of the most significant reasons for attrition in the drug discovery process (Kola and Landis, Reference Kola and Landis2004; Arrowsmith, Reference Arrowsmith2011a, Reference Arrowsmithb).
Structural information
Whilst structural information on a protein (and ideally a protein-ligand complex) is very helpful for compound optimization for target-based programmes, it is not essential. Structural information is at its most powerful when it is possible to obtain co-crystal structures of the protein with inhibitors, as this guides the design process. It can be even more helpful if there is also a structure of the human homologue (where applicable) to guide selective inhibitor design. Therefore, although structural information is not critical to target-based drug discovery, it can be an extremely powerful tool to move projects along.
PROPERTIES OF MOLECULES
In terms of drug discovery, although it is important to have a good molecular target, it is equally important to develop the molecules with the correct properties to achieve the TPP. The TPP allows criteria to be set for compounds for the different stages of the drug discovery programme in terms of potency, selectivity, pharmacokinetics and safety pharmacology. Given that during the discovery of a clinical candidate, the molecular weight and lipophilicity are likely to increase (Nadin et al. Reference Nadin, Hattotuwagama and Churcher2012), this should be taken into account when choosing the starting chemical library – compounds must be amenable to increases both in lipophilicity and molecular weight during optimization without compromising developability.
The TPPs for the kinetoplastid diseases generally require oral drugs. Lipinski defined his famous ‘rule of 5 as a guide as to whether a molecule is in the correct chemical space to be an orally bioavailable compound (Lipinski et al. Reference Lipinski, Lombardo, Dominy and Feeney1997). In his rules, compounds should have a molecular weight of less than 500, clogP <5, less than 5 hydrogen bond donors and less than 10 hydrogen bond acceptors (Lipinski et al. Reference Lipinski, Lombardo, Dominy and Feeney1997). Current thinking suggests that we should be even more stringent about the physicochemical properties of molecules. Analysis of drug molecules indicates for oral drugs, the average molecular weight is 333 and the average clogP is 2·5 (Gleeson et al. Reference Gleeson, Hersey, Montanari and Overington2011). LogP in particular appears to be correlated to a number of developability issues, including toxicity, metabolic instability and insolubility (Leeson and Springthorpe, Reference Leeson and Springthorpe2007; Hughes et al. Reference Hughes, Blagg, Price, Bailey, Decrescenzo, Devraj, Ellsworth, Fobian, Gibbs, Gilles, Greene, Huang, Krieger-Burke, Loesel, Wager, Whiteley and Zhang2008; Hann, Reference Hann2011). Recent publications suggest an ideal clogP value lies in the range of 1–3. In addition to clogP and MW, analysis indicates that other criteria are important in prospective drug candidates. (1) The 3-dimensionality is important in drug-molecules. Thus the proportion of sp3 carbon atoms in a drug is 0·47 (Lovering et al. Reference Lovering, Bikker and Humblet2009). Analysis indicates that in early drug discovery, the average proportion of sp3 carbon atoms is 0·36 and this gradually rises along the drug discovery pathway. The 3-dimensionality helps properties of molecules such as solubility by decreasing planarity and gives access to more chemical space. (2) The number of carbocyclic aromatic rings impacts negatively on the developability of molecules (Ritchie and MacDonald, Reference Ritchie and MacDonald2009; Ritchie et al. Reference Ritchie, MacDonald, Young and Pickett2011). The number of carbocyclic aromatic rings is correlated with lower solubility, higher logD, higher plasma protein binding, hERG inhibition and inhibition of some major cytochrome P450s (3A4, 2C9, 2C19). (3) For compounds required to reach the CNS, there are additional criteria. Wager et al. at Pfizer have carried out a thorough analysis of properties of compounds that have CNS penetration (Wager et al. Reference Wager, Chandrasekaran, Hou, Troutman, Verhoest, Villalobos and Will2010a, Reference Wager, Hou, Verhoest and Villalobosb). This suggested a number of ‘optimum’ values for a number of factors – for example: clogP=2·8; MW=305; Polar surface area (PSA)=45; HBD=1; pKa=8·4.
In addition to the physicochemical properties of molecules, many research laboratories filter out functional groups known to be chemically reactive or toxic (Brenk et al. Reference Brenk, Schipani, James, Krasowski, Gilbert, Frearson and Wyatt2008). In some cases this is a clear-cut decision. For example, acyl chlorides are too unstable to be developed. In other cases, the decisions about which groups to include and which to exclude are less clear. Therefore, excluding certain functional groups is a careful balancing act; excluding functional groups can make the compounds ‘cleaner’, but reduces the chemotypes covered. In some cases this becomes a judgement call, balancing the risks of certain chemical functionality against the risks of restricting the chemical space covered in a library. For example, nitro groups may be excluded due to potential genotoxicity concerns, although some drugs have aromatic nitro groups. It is important to understand the risk inherent in certain types of functional groups and to address this risk as early as possible in the discovery process, if it is decided to include these.
PROPERTIES OF CURRENT DRUGS USED FOR TREATMENT OF KINETOPLASTID INFECTIONS
Interestingly, if the current drugs used for the treatment of kinetoplastid diseases are analysed, it is clear that not many of them are ideal from a physicochemical perspective, given that the current TPPs for the kinetoplastid infections require oral drugs. Indeed most of the drugs are not orally bioavailable and require parenteral administration, as would be predicted by their physicochemical properties (Table 3 and Fig. 1). The only orally bioavailable drugs are nifurtimox, benznidazole and miltefosine. The former two are compliant with Lipinski's rules and whilst miltefosine has a logP value of >5, miltefosine is zwitterionic and the clogD value is 1·5 which is well within the Lipinski criteria. Eflornithine theoretically fulfils Lipinski's rules; however the compound is so hydrophilic (clogP=−3·7) that it is very unlikely to traverse membranes well by passive diffusion and is actually given by intravenous infusion.
Fig. 1. Current drugs used for the treatment of kinetoplastid infections.
Table 3. Physicochemical properties of currently used drugs for kinetoplastid infections

a clogP was calculated using StarDrop™.
b HDB and HBA are given as defined by Lipinski (Lipinski et al. Reference Lipinski, Lombardo, Dominy and Feeney2001).
c The precise structures of stibogluconate and meglumine antimoniate are not known.
Interestingly melarsoprol, nifurtimox and eflornithine are used for the treatment of stage 2 HAT, suggesting that the compounds are blood-brain barrier penetrants. Melarsoprol and nifurtimox are not predicted to be blood-brain barrier penetrants based on normal criteria as their polar surface areas are too high (Kelder et al. Reference Kelder, Grootenhuis, Bayada, Delbressine and Ploemen1999). Eflornithine has a marginal level of PSA, but the negative clogP and zwitterionic nature of the molecule suggest that it is not likely to be passively permeable. The uptake of these molecules is likely to be due to transporter-mediated uptake, which is very difficult to predict or plan for.
Whilst clearly there are compounds that are suitable as drug candidates that do not meet criteria such as Lipinski's rules, these mark the exception rather than the rule and the exceptions are difficult to predict as they involve factors such as transporter-mediated uptake. Therefore the physicochemical properties of molecules should be taken into account during the design process.
N-MYRISTOYLTRANSFERASE (NMT)
By way of example, in the DDU (University of Dundee), in collaboration with others, we have provided very strong validation of NMT as a drug target for HAT (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, Van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010; Brand et al. Reference Brand, Cleghorn, McElroy, Robinson, Smith, Hallyburton, Harrison, Norcross, Spinks, Bayliss, Norval, Stojanovski, Torrie, Frearson, Brenk, Fairlamb, Ferguson, Read, Wyatt and Gilbert2012). I will briefly summarize its validation process below.
NMT is an enzyme found in all eukaryotic cells; its function is co-translational modification of a number of proteins, by N-myristoylation of their N-terminal glycine atoms. NMT has been investigated as a potential target for fungal diseases by a number of companies; but, despite discovery of some very potent inhibitors, none of these have been progressed to market.
Within T. brucei, NMT has been genetically validated initially by RNAi experiments by Price et al. (Reference Price, Menon, Panethymitaki, Goulding, McKean and Smith2003), and then kinetically characterized by Panethymitaki et al. (Reference Panethymitaki, Bowyer, Price, Leatherbarrow, Brown and Smith2006) in York. We set about validation of NMT as a target in T. brucei. It was then established that when NMT is knocked-down by RNAi, the parasites are unable to establish an infection in mice, adding further genetic validation (Price et al. Reference Price, Guther, Ferguson and Smith2010). To obtain chemical validation, at the DDU, we profiled a number of reported inhibitors of fungal NMT against T. brucei NMT (Brand et al. Reference Brand, Cleghorn, McElroy, Robinson, Smith, Hallyburton, Harrison, Norcross, Spinks, Bayliss, Norval, Stojanovski, Torrie, Frearson, Brenk, Fairlamb, Ferguson, Read, Wyatt and Gilbert2012); but none of these showed significant inhibition of the T. brucei enzyme. We therefore carried out a high-throughput screen using a 62 000 compound diversity library (Brenk et al. Reference Brenk, Schipani, James, Krasowski, Gilbert, Frearson and Wyatt2008) from the DDU. This led to identification of a series of pyrazole sulphonamides as hit molecules, with the most potent compound having an IC50 value of 1·9 μ m (Fig. 2). These were optimized to a potent inhibitor, DDD85646 with an IC50 value of 0·002 μ m against T. brucei NMT and EC50 value of 0·002 μ m against the parasite. We were then able to show that the compounds were active in rodent models of infection, both for T. b. brucei (curative at 12·5 mg/kg po bid for 4 days) and T. b. rhodesiense (curative at 50 mg/kg po bid for 4 days).
Fig. 2. Compounds active against T. brucei NMT.
In order to validate that these compounds were acting against NMT, a number of experiments were undertaken. These have been reported previously (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, Van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010), but are summarized here to show how they are important in validation of the target. (1) It was possible to obtain a co-crystal structure of the inhibitor DDD85646 with the T. brucei NMT homologue, Leishmania major NMT. This shows the inhibitor bound in the enzyme active site, occupying the peptide binding pocket. This is evidence that the compound can bind to the enzyme and that it binds in the substrate binding pocket. (2) We looked at a large number of inhibitors of T. brucei NMT and compared them to inhibition of parasite growth. There was a very good correlation between inhibition of the enzyme and inhibition of parasite growth, which is a good indication that inhibition of TbNMT is responsible for the death of the parasites. (3) The gene encoding for NMT was over-expressed within the parasite T. brucei by about 5-fold. This gave rise to an 8-fold change in potency from 2·1 to 17 nm. This indicates that the compounds are inhibiting NMT and that cell death is due to inhibition of NMT. (4) A further experiment was to incubate the parasites with radio-labelled myristic acid. This is incorporated into proteins through myristoylation, which could be checked by SDS-PAGE. When the experiment was repeated in the presence of DDD85646, myristoylation is prevented and no radio-labelled proteins could be detected. This is good evidence that in cells, NMT inhibitors prevent myristoylation of proteins.
Therefore, through a suite of experiments, we have obtained good evidence that NMT is a valid drug target for HAT (Frearson et al. Reference Frearson, Brand, McElroy, Cleghorn, Smid, Stojanovski, Price, Guther, Torrie, Robinson, Hallyburton, Mpamhanga, Brannigan, Wilkinson, Hodgkinson, Hui, Qiu, Raimi, Van Aalten, Brenk, Gilbert, Read, Fairlamb, Ferguson, Smith and Wyatt2010). The target has been genetically validated through RNAi both in cells and in a mouse model of infection. We have demonstrated that our compounds inhibit NMT through binding to the peptide binding pocket of the enzyme. Inhibition of NMT prevents myristoylation of proteins in cells and leads to cell death of T. brucei. When dosed to mice orally, at sufficient levels to give unbound blood levels at EC99, these NMT inhibitors cure animal models of HAT.
In terms of physicochemical properties, these compounds are appropriate for oral bioavailability: i.e. MW=495; cLogP=3·1; HBD=2; HBA=5. However, DDD85646 does not have the correct physicochemical properties for CNS penetration, and indeed it is not active in a CNS model of infection. Interestingly when we started this project, there was a TPP for oral stage 1 compounds for HAT but during the work on this project, the priority was changed to requiring a TPP activity in the CNS stage 2 of infection. This demonstrates how TPPs can alter and the effect that they can have on the types of molecule that are required for the indication.
CONCLUSION
Target-based drug discovery is possible for HAT but careful selection of the molecular target must be made. Often there is insufficient information available to be certain of how valid selected molecular targets are at the beginning of a drug discovery project. It is therefore important that the correct biological experiments are carried out and tool molecules prepared to validate the molecular targets early in the project. These include genetic validation experiments and mode of action experiments with tool molecules to demonstrate that the tool molecules are acting on target and it is through inhibition of the target that they exert their anti-parasitic activity. Thought must also be put into selection of compounds for screening and the compounds that are developed, to make sure that they have the correct physicochemical and pharmacokinetic properties.
ACKNOWLEDGEMENTS
I would like to acknowledge members of the Drug Discovery Unit at the University of Dundee (UK) for contributions to this HAT project and discussions around target validation and compound selection.
FINANCIAL SUPPORT
The NMT project in the Drug Discovery Unit was funded by the Wellcome Trust (WT077705 and WT083481).





 Open access
Open access