



Introduction
Scathophaga stercoraria (yellow dung fly) is a species of fly in Scathophagidae, which is a small family of Muscoidea (Vockeroth, Reference Vockeroth1987). Most species of Scathophagidae are distributed in the Holarctic, mostly in northern latitudes (Mortelmans and Devillers, Reference Mortelmans and Devillers2014). S. stercoraria is widespread and typically seen on the dung of large mammals, especially on cattle dung (Blanckenhorn, Reference Blanckenhorn1998). S. stercoraria attracted early attention as a possible biocontrol agent (Cotterell, Reference Cotterell1920). Meanwhile, this fly is also a popular subject for many other studies, including sperm competition (Gress et al., Reference Gress, Starmer, Virgen, Agu, Attila, Bazluke and Pitnick2016), sexual selection (Sbilordo et al., Reference Sbilordo, Martin and Ward2010), reproductive physiology (Reim et al., Reference Reim, Teuschl and Blanckenhorn2006), growth and development (Hosken et al., Reference Hosken, Blanckenhorn and Ward2000; Walters et al., Reference Walters, Berger, Blanckenhorn, Bussière, Rohner, Jochmann and Schäfer2022), genetic aspects (Demont et al., Reference Demont, Blanckenhorn, Hosken and Garner2008), thermobiology (Blanckenhorn et al., Reference Blanckenhorn, Berger, Rohner, Schäfer, Akashi and Walters2021), immune-related research (West and Tracy, Reference West and Tracy2009). In addition, S. stercoraria is often used for ecotoxicological tests, such as ivermectin detection (Mahdjoub et al., Reference Mahdjoub, Blanckenhorn, Lüpold, Roy, Gourgoulianni and Khelifa2020; González-Tokman et al., Reference González-Tokman, Bauerfeind, Schäfer, Walters, Berger and Blanckenhorn2022).
The plateau pika (Ochotona curzoniae) (Lagomorpha, Ochotonidae) is a specialized native species of the Qinghai-Tibetan plateau (QTP) (Zhu et al., Reference Zhu, Zhong, Li, Wang and Qu2022). It is important to the community of grasslands in the QTP (Zhang et al., Reference Zhang, Qu, Li, Li, Yang and Zhang2017). The digging activities of the plateau pika increase the abundance of plant species (Qin et al., Reference Qin, Huang, Zhang, Yu, Yi and Sun2021) and the dug burrows can become habitats for other animals (lizards, and small birds) (Zhao et al., Reference Zhao, Zhao, Xu and Xu2020). Previous studies have shown that plateau pikas are susceptible to infestation by various parasites, including Oestromyia leporina (Fu et al., Reference Fu, Li, Duo, Guo, Dang, Shen, Peng and Zhang2016), Echinococcus multilocularis (Li et al., Reference Li, Li, Fan, Fu, Zhu, Yan and Jia2018), Taenia spp. (Wu et al., Reference Wu, Li, Fan, Ni, Ohiolei, Li, Li, Zhang, Fu, Yan and Jia2021), Cryptosporidium spp. (Zhang et al., Reference Zhang, Jian, Li, Ma, Karanis and Karanis2018), Toxoplasma gondii (Zhang et al., Reference Zhang, Lou, Huang, Zhou, Jia, Su and Zhu2013), Enterocytozoon bieneusi (Liu et al., Reference Liu, Du, Yang, Xia, An and Qi2021). However, the presence of S. stercoraria in plateau pikas has never been reported before.
The QTP and its surrounding mountain systems are one of the hotspots of biodiversity (Wu et al., Reference Wu, Dai, Littlewood, Ohiolei, Guo, Shumuye and Jia2022) and have played important roles in the evolution of organisms (Rahbek et al., Reference Rahbek, Borregaard, Antonelli, Colwell, Holt, Nogues-Bravo, Rasmussen, Richardson, Rosing, Whittaker and Fjeldså2019). The uplift of the QTP and associated climate change has driven species diversity on the plateau (Mao et al., Reference Mao, Wang and Liu2021), while also causing the isolation and divergence of many species (Flantua et al., Reference Flantua, O'Dea, Onstein, Giraldo and Hooghiemstra2019; Rahbek et al., Reference Rahbek, Borregaard, Antonelli, Colwell, Holt, Nogues-Bravo, Rasmussen, Richardson, Rosing, Whittaker and Fjeldså2019). The mitochondrial genome is characterized by low molecular weight and genetic conservation due to its largely haploid and uniparentally inherited feature. Thus, in recent years, the mitochondrial genome has been widely used in the study of phylogenetic relationships, molecular evolution and population genetics (Pyziel et al., Reference Pyziel, Laskowski, Dolka, Kołodziej-Sobocińska, Nowakowska, Klich, Bielecki, Żygowska, Moazzami, Anusz and Höglund2020).
In this study, the complete mitochondrial genome of S. stercoraria was sequenced and annotated, the phylogenetic tree was reconstructed using 40 mitochondrial genomes and complemented by phylogenetic trees generated on 18S rDNA, 28S rDNA and COI gene. The possible divergence time of Scathophaga in history was investigated by the COI gene. These studies have provided fundamental data to better understand the phylogenetic relationships and evolutionary history of S. stercoraria in the QTP area.
Materials and methods
Sample collection and DNA extraction, PCR, sequencing
Five larvae were collected subcutaneously from the hind limbs of plateau pika in Chenduo county (33°35′N; 97°12′E; altitude at 4377 m) of Qinghai Province, the People's Republic of China in June 2022. After being washed in phosphate saline buffer, all samples were stored in 70% ethanol. Genomic DNA were extracted from the samples using a commercial kit (TIANamp Genomic DNA Kit, TIANGEN Biotechnology, Beijing, China) according to the manufacturer's instructions. Partial sequences of COI (Otranto et al., Reference Otranto, Colwell, Traversa and Stevens2003), 18S rDNA (Nirmala et al., Reference Nirmala, Hypsa and Zurovec2001) and 28S rDNA (Otranto et al., Reference Otranto, Colwell and Pape2005) were determined using primers that have been previously reported. Primers were synthesized by Sangon Biotech (Shanghai, China), standard 25 μL PCR protocol was used to amplify the DNA fragments. The PCR products were purified using a TIANgel Midi Purification Kit (Cat. DP209-02, Tiangen, China), and finally sent to Sangon Biotech (Shanghai) Co., Ltd. for sequencing. The list of primers and PCR reaction conditions are shown in Supplementary Table 1.
Mitochondrial genome sequencing, assembly and annotation
DNA samples were sent to Sangon Biotech (Shanghai) Co., Ltd. for library construction and sequencing. Library construction using a whole genome shotgun (WGS) strategy was performed, followed by next-generation sequencing to obtain mitochondrial genome sequences. For the quality-checked fragments, sequencing was performed on the Illumina Hiseq platform using a double-end sequencing strategy. Low quality sequences were removed from raw sequences with FASTP v0.36 (Chen et al., Reference Chen, Zhou, Chen and Gu2018) software to obtain a clean data dataset. SPAdes v3.15 (Bankevich et al., Reference Bankevich, Nurk, Antipov, Gurevich, Dvorkin, Kulikov, Lesin, Nikolenko, Pham, Prjibelski, Pyshkin, Sirotkin, Vyahhi, Tesler, Alekseyev and Pevzner2012) software was used to splice and assemble the short fragment sequences (Clean reads) from high-throughput sequencing. After the assembly was completed, assembled sequences were compared with the known S. stercoraria genes in Gen Bank. Sequencing results were subsequently confirmed as the S. stercoraria mitochondria genome. The complete mitochondrial genome sequence of S. stercoraria was successfully obtained. The 37 genes of the S. stercoraria mitochondrial genome were annotated by the online software MITOS Web Server (http://mitos.bioinf.uni-leipzig.de/index.py) to determine the position of each gene and predict the secondary structure of tRNA (Bernt et al., Reference Bernt, Donath, Jühling, Externbrink, Florentz, Fritzsch, Pütz, Middendorf and Stadler2013). The annotation results were exported using SnapGene v7.0 software and manually corrected by referring to the reported mitochondrial genome annotation results of S. stercoraria in NCBI.
Phylogenetic analyses
To determine the phylogenetic relationships of S. stercoraria, 13 Protein coding genes (13PCGs), 18S and 28S rDNA, COI gene in this study with those of other classified Diptera available in GenBank were used as ingroup, as 2 species (Batocera horsfieldi, Trigoniopthalmus alternatus) of insects belonging to different orders were chosen as outgroups. To analyse phylogenetic relationships, 4 datasets were collected which containing 108 species from 6 families (with evolutionary trees of 18S and 28S rDNA as a complement to the evolutionary tree of 13PCGs); due to little has been previously reported about the phylogeny of the genus Scathophaga with other species within the family Scathophagidae, 37 species were selected from 18 genera in the family Scathophagidae and the phylogenetic relationships were analysed (see Supplementary Tables 2–5 for details). Owing to limited data on different genes of the same species, the phylogeny of each dataset was performed independently. Sequence alignment was accomplished using the MAFFT v7.505 with auto option (Katoh and Standley, Reference Katoh and Standley2013). TrimAl v1.2 was used under the automated 1 option to trim the aligned sequences (Capella-Gutiérrez et al., Reference Capella-Gutiérrez, Silla-Martínez and Gabaldón2009). The phylogenetic tree was constructed using the maximum likelihood (ML) method with IQ-TREE v2.2.0 (Nguyen et al., Reference Nguyen, Schmidt, von Haeseler and Minh2015). The selection of models is performed automatically by ModelFinder (Kalyaanamoorthy et al., Reference Kalyaanamoorthy, Minh, Wong, Von Haeseler and Jermiin2017). All other parameters were set to the default values. The online tool: tvBOT (Xie et al., Reference Xie, Chen, Cai, Cai, Hu and Wang2023) was used to view and modify phylogenetic trees.
Divergence times estimates
Due to the lack of a complete mitochondrial genome for other Scathophaga species, the COI gene was used to analyse the divergence time. Divergence times were estimated using BEAST v2.7.4 (Bouckaert et al., Reference Bouckaert, Heled, Kühnert, Vaughan, Wu, Xie, Suchard, Rambaut and Drummond2014); the clock model was set to relaxed, uncorrelated log-normal and the gamma category count was set to 4; and the GTR substitution model was selected. For the tree prior the Calibrated Yule model (Heled and Drummond, Reference Heled and Drummond2015) was used. Due to the lack of Scathophaga fossils, a secondary calibration approach was used (Hedges and Kumar, Reference Hedges and Kumar2004). Based on previous research on divergence time in Scathophaga (Junqueira et al., Reference Junqueira, Azeredo-Espin, Paulo, Marinho, Tomsho, Drautz-Moses and Schuster2016), the time calibration was set as 41 million years ago (Mya). The posterior probability estimates were drawn every 1000 steps out of the total 10 000 000 steps of each MCMC run. Other options were run on the default values. Tracer (v1.7.2) was used to determine whether the result converges. TreeAnnotator (v2.1.2) was used to annotate the tree by using maximum clade credibility tree and median heights settings with 10% burn-in.
Results
Characterization of the mitochondrial genome
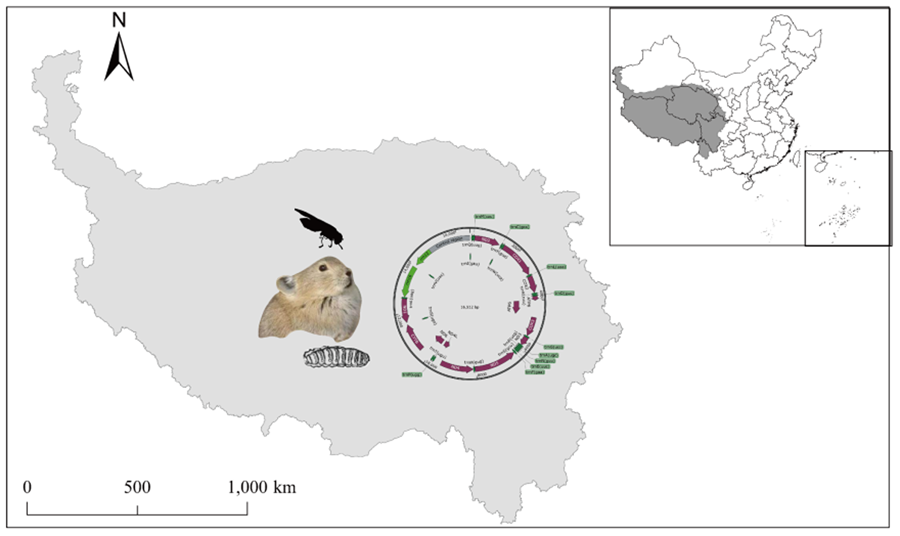
The total length of the mitochondrial genome in S. stercoraria was determined to be 16 512 bp (GenBank ID: OR039275), consisting of circular DNA molecules. The mitochondrial genome exhibits features typical of insect mitochondrial genomes; double-stranded DNA molecules, including light-stranded L and heavy-stranded H, 2 rRNAs: 16s rRNA, 12s rRNA, 22 tRNAs, 13PCGs and a large control region (D-Loop region) (Fig. 1). The inferred gene boundaries and lengths are shown in Table 1. Our findings are consistent with previous reports (Li et al., Reference Li, Wang, Su and Yang2016).

Figure 1. The diagram of complete mitochondrial genome of S. stercoraria. The mitochondrial genome consists of protein-encoding genes (plum), tRNAs (green), rRNAs (light green) and non-coding mitochondrial regions (Control region) (grey). The inferred gene boundaries of them are shown in Table 1.
Table 1. The list of mitochondrial genome annotation for Scathophage stercoraria

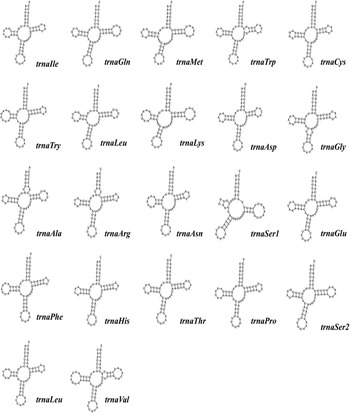
The standard start codons of invertebrate PCGs include ATN, GTG and TTG (Wolstenholme, Reference Wolstenholme1992a, Reference Wolstenholme1992b). Except for CO1, which uses TCG in S. stercoraria, all PCGs start with standard start codons. Other Diptera mitochondrial genomes frequently contain these unconventional start codons. Twelve PCGs of S. stercoraria terminate with the common stop codons TAA or TAG, except for the NAD4, which ends with a single thymine stop codon (Table 1). Incomplete stop codons are hypothesized to be filled by polyadenylation during mRNA maturation (Ojala et al., Reference Ojala, Montoya and Attardi1981). All tRNAs can be folded into typical cloverleaf structures, except trnSer1 for its DHU arm, forming a simple loop (Fig. 2), and this has been repeatedly reported in other metazoan mitochondrial genomes (Wolstenholme, Reference Wolstenholme1992a, Reference Wolstenholme1992b).

Figure 2. The structure of 22 tRNAs derived from the S. stercoraria mitochondrial genome. Structures of 22 tRNAs with base pairs are shown, with the names of the tRNAs and anticodons displayed in the bottom right of each structure.
Phylogenetic relationships
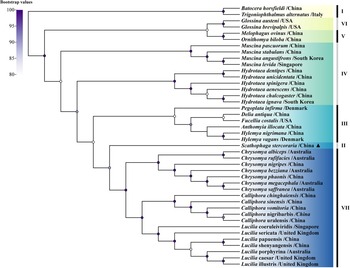
The result of phylogenetic analysis constructed based on the sequences of 13PCGs showed that S. stercoraria has a close genetic affinity with the family Calliphoridae (Fig. 3), the same results were also obtained from the analysis of 18S and 28S rDNA (Fig. 4). Phylogenetic analysis based on COI gene showed that S. stercoraria itself, as a monophyletic group, has a close genetic affinity with Leptopa filiformis, Micropselapha filiformis, Parallelomma medium, Parallelomma paridis and Americina adusta (Fig. 5).

Figure 3. Maximum likelihood analyses of S. stercoraria based on 13PCGs. The different coloured bars and the Roman numerals to the right represent outgroups and different genus names (i.e. I: outgroup, II: Scathophagidae, III: Anthomyiidae, IV: Muscidae, V: Hippoboscidae, VI: Glossinidae, VII: Calliphoridae). ▴ was used to mark the S. stercoraria of this study. Nodes with Bootstrap values > 80% are displayed.

Figure 4. Maximum likelihood analyses of S. stercoraria based on 18S rDNA (a) and 28S rDNA (b) fragments. The different coloured bars and the Roman numerals to the right represent outgroups and different genus names (i.e. I: outgroup, II: Scathophagidae, III: Anthomyiidae, IV: Muscidae, V: Hippoboscidae, VI: Glossinidae, VII: Calliphoridae). ▴ was used to mark the S. stercoraria of this study. Nodes with Bootstrap values > 80% are displayed.

Figure 5. Maximum likelihood analyses of S. stercoraria based on COI fragments. S. stercoraria of this study is marked with ▴ and red. Nodes with Bootstrap values > 80% are displayed.
Divergence times analysis
The divergence time analysis based on the COI gene suggested that the most recent common ancestor of the twenty Scathophaga species existed at approximately 40.98 Mya, this is in accordance with previous reports (Junqueira et al., Reference Junqueira, Azeredo-Espin, Paulo, Marinho, Tomsho, Drautz-Moses and Schuster2016). The divergence time between S. stercoraria and the other twelve Scathophaga species was approximately 27.07 Mya, and the divergence time of the sixteen Scathophaga species all occurred within 1 Mya (Fig. 6).

Figure 6. Divergence time of S. stercoraria was estimated based on COI fragments. ▴ was used to mark the S. stercoraria of this study. The green bar represents an interval of 95% highest probability density. Different colours indicate different periods (Eocene, Oligocene, Miocene, Pliocene and Pleistocene).
Discussion
The presence of S. stercoraria in pika is related to yak feces
This is the first documentation of S. stercoraria collected in plateau pikas and the first report of the complete mitochondrial genome of S. stercoraria in the QTP. According to previous reports, S. stercoraria generally reproduces on dung and their larvae grow in the dung (Gress et al., Reference Gress, Starmer, Virgen, Agu, Attila, Bazluke and Pitnick2016). However, a new discovery has been made: the presence of S. sterocoraria in plateau pikas. As S. stercoraria has only been previously reported to be present in the dung of animals with no reports showing its presence in the animal itself, based on this, it was speculated that the presence of S. stercoraria in plateau pikas is possibly attributed to the contamination from the dung of yaks or other large mammals during their daily activities. Female S. stercoraria are accustomed to laying eggs on the surface of dung to avoid the living environment of eggs being too dry or too humid (Ward et al., Reference Ward, Foglia and Blanckenhorn1999). This creates conditions for plateau pikas to carry S. stercoraria eggs from the dung of large mammals. After the eggs are transferred from dung to the plateau pika, the environment where the eggs are located may not provide adequate conditions for their continued growth and development. Therefore, in order to survive, the eggs may have burrowed into the subcutaneous tissue of the plateau pika after developing into a larva.
Based on the above reasons, it was speculated that plateau pika carries the eggs of S. stercoraria from the dung of yaks. Study shows that the 2 species began to coexist at about 2.4 Mya and they compete for phytophagous food and overlapping spaces (Harris et al., Reference Harris, Wenying, Badinqiuying, Smith and Bedunah2015). In addition, it was found that during winter, when food is scarce, plateau pikas survive by ingesting yak feces (Speakman et al., Reference Speakman, Chi, Ołdakowski, Fu, Fletcher, Hambly, Togo, Liu, Piertney, Wang, Zhang, Redman, Wang, Tang, Li, Cui, Thomson, Wang, Glover, Robertson and Wang2021) and develop reciprocal relationships through horizontal transmission of the gut microbiota (Fu et al., Reference Fu, Zhang, Fan, Li, Liu, Zhang and Zhang2021). Meanwhile, S. stercoraria is thought to occur more often in cattle dung (Blanckenhorn et al., Reference Blanckenhorn, Pemberton, Bussière, Roembke and Floate2010). Therefore, it was speculated that the plateau pika carries the eggs of S. stercoraria from the yak dung, and the larvae parasitize in plateau pikas in order to survive. As this is the first discovery of S. stercoraria ‘parasitism’ in animals, further study is needed to determine whether this parasitic behaviour of S. stercoraria is an accidental event or an adaptive change made to adapt to the harsh living environment of the QTP.
Mitochondrial genomic characterization and phylogenetic analysis
The complete mitochondrial genome of S. stercoraria was sequenced and annotated, and the sequence was similar to the reported mitochondrial whole genome of S. stercoraria (Li et al., Reference Li, Wang, Su and Yang2016). They share similarities in the rRNA, tRNA and protein-encoding genes regarding length, gene order and composition. The difference between the two lies in the control region (D-Loop region). The control region in this study was approximately 400 bp longer than that from previous studies. The differences are potentially attributed to the fact that S. stercoraria collected in our study versus the S. stercoraria from previous studies were collected in different places, resulting in differences in their adaptation to the local environment. The phylogenetic tree shows that S. stercoraria is more closely related to Lucilia, Calliphora and Chrysomya of the family Calliphoridae, which is consistent with previous findings (Ding et al., Reference Ding, Li, Wang, Cameron, Mao, Wang, Xi and Yang2015). The phylogenetic tree obtained based on the COI gene within the family Scathophagidae shows that S. stercoraria is closely related to the genus Leptopa, Micropselapha, Parallelomma and Americina. This adds to the previously reported phylogenetic relationships within the Scathophagidae family; in addition, it was found that the affinities of S. stercoraria and Americina adusta in this study were inconsistent with previously reported studies (Bernasconi et al., Reference Bernasconi, Pawlowski, Valsangiacomo, Piffaretti and Ward2000a, Reference Bernasconi, Valsangiacomo, Piffaretti and Ward2000b), and it was speculated that this is potentially due to the location of sample collection (S. stercoraria in Bernasconi's study was from Switzerland) and the selection of different outgroups.
Divergence time and evolution of the Scathophaga
The evolution, differentiation, or diversity of organisms may be influenced by biotic factors such as competition, intraspecific interactions, and abiotic factors such as tectonic events and climate, or by the combination of both (Antonelli and Sanmartín, Reference Antonelli and Sanmartín2011). Biotic factors tend to influence organisms over a short period (less than 1 Mya), while abiotic factors drive evolutionary differentiation over a longer period (millions of years or even longer) (Benton, Reference Benton2009). Because the events associated with the uplift of the QTP span tens of millions of years, the effects of biotic factors on organisms during this process are likely to be lower than those of abiotic factors (Favre et al., Reference Favre, Päckert, Pauls, Jähnig, Uhl, Michalak and Muellner-Riehl2015). Our divergence time analysis based on the COI gene estimated that there are 2 important divergence times for the genus Scathophaga, 27 and 20 Mya, and except for S. suilla, S. soror, S. apicalis, S. stercoraria, the rest of Scathophaga underwent divergence at approximately 1 Mya. This indicates that the geological and climatic events during these periods (late Oligocene to early Miocene, Pliocene, Pleistocene) (Harrison et al., Reference Harrison, Copeland, Kidd and Yin1992; Ni et al., Reference Ni, Li, Li and Beard2016) may have played an important role in the differentiation of Scathophaga.
The uplift of the QTP has caused environmental and climatic changes that have driven the evolution of associated biotas (Favre et al., Reference Favre, Päckert, Pauls, Jähnig, Uhl, Michalak and Muellner-Riehl2015). During the Oligocene and Miocene periods, the uplift of the QTP advanced to the north and south, which caused the extension of the QTP (35 to 20 Mya) (Mulch and Chamberlain, Reference Mulch and Chamberlain2006; Qiang et al., Reference Qiang, An, Song, Chang, Sun, Liu and Ai2011; Miao et al., Reference Miao, Herrmann, Wu, Yan and Yang2012). The carbon dioxide content in the atmosphere during the Oligocene was lower than that during the Eocene, which resulted in a warmer global climate during the Oligocene (Villa and Persico, Reference Villa and Persico2006; O'Brien et al., Reference O'Brien, Huber, Thomas, Pagani, Super, Elder and Hull2020). During the Miocene period, orogenic movements in high mountain ranges, such as the Himalayas led to the gradual expansion of the uplift of the QTP (Tapponnier et al., Reference Tapponnier, Zhiqin, Roger, Meyer, Arnaud, Wittlinger and Jingsui2001; Wang et al., Reference Wang, Zhao, Liu, Lippert, Graham, Coe, Yi, Zhu, Liu and Li2008). In addition, paleobotanical data indicates that the southeastern edge of the QTP was dominated by a warm and humid climate during the Miocene period, primarily influenced by the monsoon winds from East and South Asia (Sun and Wang, Reference Sun and Wang2005; Jacques et al., Reference Jacques, Guo, Su, Xing, Huang, Liu and Zhou2011). The Earth's climate underwent a fundamental change during the Pleistocene, known as the Middle Pleistocene Transition (MPT); during this time, the climate changed more and more drastically, and the Northern Hemisphere became increasingly glaciated (Pena and Goldstein, Reference Pena and Goldstein2014; Sun et al., Reference Sun, Yin, Crucifix, Clemens, Araya-Melo, Liu and An2019).
The timeline of geological and climate change mentioned above is close to the timeline of Scathophaga differentiation obtained in this study (Fig. 4). It was speculated that the uplift of the QTP, along with global climate change, drove the divergence of the Scathophaga genus. The complex mountainous regions of western China (including the QTP, the Himalayas, the Hengduan Mountains, and the Three Gorges Mountains) are responsible for the isolation and divergence of many plants and animals (Yuan et al., Reference Yuan, Zhang, Peng and Ge2008; Zhang et al., Reference Zhang, Rao, Yang, Yu and Wilkinson2010). During the Oligocene and Miocene periods, the uplift of the QTP and the resulting climate change drove the formation of species and the diversity of their populations in western China (He et al., Reference He, Cao and Chen2001; Che et al., Reference Che, Zhou, Hu, Yan, Papenfuss, Wake and Zhang2010); and the species of Scathophaga gradually differentiated. In addition, the warm and humid climate of the Miocene period created a suitable environment for the development of biodiversity (Barry et al., Reference Barry, Morgan, Flynn, Pilbeam, Behrensmeyer, Raza and Kelley2002; Badgley et al., Reference Badgley, Barry, Morgan, Nelson, Behrensmeyer, Cerling and Pilbeam2008; LaRiviere et al., Reference LaRiviere, Ravelo, Crimmins, Dekens, Ford, Lyle and Wara2012), which accelerated the differentiation of the Scathophaga genus. Finally, the drastic climate changes during the Pleistocene period resulted in ecological variability, which in turn affected all aspects of flora and fauna (Hofreiter and Stewart, Reference Hofreiter and Stewart2009); this may also be the reason why most Scathophaga species differentiated and formed during the Pleistocene. Therefore, it was inferred that climate change during the Pleistocene and the uplift of the QTP are the 2 most important factors influencing Scathophaga differentiation.
Conclusion
In conclusion, in this study, the phylogenetic analysis of S. stercoraria was conducted for the first time using mitochondrial genes, 18S rDNA and 28S rDNA. Additionally, the divergence time of Scathophaga was estimated for the first time using the COI gene. It was suggested that S. stercoraria in plateau pikas may be derived from yak feces, and S. stercoraria was differentiated and formed around the early Miocene (21 Mya) due to the uplift of the QTP and climate change. This study provides fundamental information for the subsequent study of the kinship and differentiation of S. stercoraria. However, due to the lack of reports on S. stercoraria in the QTP, more samples need to be collected to further study the growth and development of S. stercoraria in the QTP and to elucidate the phylogeny and differentiation of Scathophaga in more detail.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182024000623
Data availability
Data will be made available on request.
Author contributions
Yong Fu, Donglei Sun and Zhi Li: Conceptualization, Project administration, Data analysis, Manuscript drafting, Review and revision. Haining Zhang: Executing laboratory experiments, Manuscript drafting. Wangkai Chen: Executing laboratory experiments, Data analysis. Ru Meng and Hong Duo: Conceptualization, Data analysis, Review & Editing. Xueyong Zhang, Zhihong Guo, Xiuying Shen and Qing Liu: Data analysis. All authors read and approved the final manuscript.
Financial support
This work was supported by grants from the Applied Basic Research of Qinghai Province in China (Grant No. 2021-ZJ-724), the National Science Foundation (32100723), Shanghai Municipal Natural Science Foundation (21ZR1432600) to DS, and the Open Project of State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University (Grant No. 2022-ZZ-10).
Competing interest
The authors declared that they have no conflicts of interest to this work.
Ethical standards
Not applicable.








 Open access
Open access