Introduction
Nutrition in early life is crucial for fetal growth and development. Food intake absence and excess are the two main types of energy malnutrition that predispose to the appearance of diseases in adulthood, according to the hypothesis of ‘developmental origins of health and disease’( Reference Barker 1 ). In this regard, the transmission of an altered metabolic phenotype to the progeny can lead to an increased risk of developing different metabolic diseases, including non-alcoholic fatty liver disease (NAFLD).
NAFLD results from the interaction between environmental and genetic factors. Maternal diet constitutes a major environmental factor which directly affects the liver and indirectly through adiposity and metabolic dysfunction. Different studies support the notion that early-life malnutrition may affect the metabolic phenotype in the offspring, leading to the development of NAFLD( Reference Brumbaugh and Friedman 2 ). In addition, evidence has been obtained showing that epigenetics exerts an important role in NAFLD pathogenesis( Reference Gallego-Durán and Romero-Gómez 3 ). These epigenetic mechanisms are considered a link between the nutritional environment and gene expression. Increasing evidence has shown that early-life nutrition can influence the development of the immune system through epigenetic mechanisms( Reference Paparo, di Costanzo and di Scala 4 ). Thus, epigenetics has a crucial effect over the intricate interplay between environmental factors and genetics to drive the inflammatory response that is commonly seen in NAFLD.
The aim of the present review is to describe the epigenetic modulation of the immune function and its association with the development of liver diseases related to early-life malnutrition.
Role of early-life malnutrition in the developmental origin of non-alcoholic fatty liver diseases
Malnutrition is determined as a disequilibrium between energy intake and the quantity that the body needs for optimal growth and function. There are many causes of malnutrition such as obesity, protein–energy malnutrition, food intake absence and essential fatty acid deficiency among others, although food intake absence and excess are the two main types of energy malnutrition( Reference Singer, Stancu and Coşoveanu 5 ). Since the nutritional environment during preconception, pregnancy and early life is crucial for optimal offspring development and long-term health, it has important consequences for survival, healthy development and the incidence of acute and chronic diseases in later life( Reference Black, Victora and Walker 6 ). Certainly, it has been proposed that the period between pregnancy and the first 2 years of life is critical. In this respect, it has been shown that chronic diseases are especially common in undernourished children who experience rapid weight gain after infancy( Reference Victora, Adair and Fall 7 ). The ‘thrifty phenotype’ hypothesis( Reference Hales and Barker 8 ) explains this phenomenon, arguing that fetal undernutrition leads to metabolic adaptations that optimise the utilisation of limited nutrient availability and, hence, augment the opportunities of survival in continued poor conditions after birth. Nevertheless, these adaptations increase the risk for metabolic alterations when exposed to a plentiful postnatal nutrient environment( Reference Gluckman and Hanson 9 ). In this respect, the phenomenon of fetal intra-uterine growth restriction can occur in humans as a result of poor maternal nutrition and has been associated with the development of different metabolic diseases, a process called fetal programming( Reference Barker, Eriksson and Forsén 10 ). Epidemiological investigations such as those conducted on children conceived during the Dutch ‘Hunger Winter’ of 1944–1945 or during the great Chinese famine of 1950–1960 highlighted the association between maternal undernutrition, lowered birth weight, and subsequent adult diseases such as obesity, diabetes, hyperlipidaemia, NAFLD and even the metabolic syndrome( Reference Ravelli, van Der Meulen and Osmond 11 – Reference Ross and Beall 13 ). On the other side, different studies on diabetic pregnancies and maternal obesity demonstrated that an excessive energy intake during gestation and childhood has similar effects on offspring long-term health outcomes( Reference Pettitt, Baird and Aleck 14 – Reference Whitaker 17 ). Obesity programmes the fetus in a similar way as maternal undernutrition. This association of maladaptive programming with adult diseases has generated a great attention to the developmental programming process. Since NAFLD has an elevated prevalence and commonly an unfavourable evolution, it represents an important public health care issue( Reference Pardee, Lavine and Schwimmer 18 ).
Epidemiological characteristics of non-alcoholic fatty liver disease
The epidemiological association between early-life malnutrition and the metabolic syndrome in later life has been well documented( Reference Ravelli, van Der Meulen and Osmond 11 – Reference Ross and Beall 13 , Reference Correia-Branco, Keating and Martel 19 – Reference Fraser, Ebrahim and Smith 21 ). NAFLD is the hepatic manifestation of the metabolic syndrome and its diagnosis is strongly predictive of other features of this mentioned syndrome that include insulin resistance, obesity, diabetes, hyperlipidaemia and hypertension( Reference Ravelli, van Der Meulen and Osmond 11 – Reference Ross and Beall 13 , Reference Correia-Branco, Keating and Martel 19 – Reference Cianfarani, Agostoni and Bedogni 24 ). NAFLD is the most common cause of chronic disease around the world, both in adults and children, with a prevalence ranging between 20 and 30 % in Western countries( Reference Dietrich and Hellerbrand 25 ). It covers a spectrum of diseases ranging from steatosis or a simple accumulation of fat that exceeds 5 % of total liver weight, to non-alcoholic steatohepatitis (NASH) that also involves inflammation and significant hepatocyte cell death. Because NAFLD can evolve to liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC), which do not have effective treatments until today, it is predicted to become the most frequent indication for liver transplantation by 2030( Reference Byrne and Targher 26 ). NAFLD has a prevalence estimated in children that ranges from 3 to 10 % in Western societies and in adolescents prevalence has more than doubled in the last two decades( Reference Brumbaugh and Friedman 2 ). Interestingly, the prevalence of NAFLD is different between ethnic groups. Africans Americans are protected from hepatic steatosis compared with Caucasian and Hispanic adults, even when their BMI and insulin sensitivity are controlled for( Reference Brumbaugh and Friedman 2 ). It is important to note that most cases of NASH are found in adults rather than in children and, like other chronic inflammatory processes in the liver, progression of NASH to clinically relevant hepatic fibrosis usually takes several years.
Non-alcoholic fatty liver disease: its contribution to the risk of developing hepatocellular carcinoma
As has been previously exposed, NAFLD is a growing health problem. Patients go through a wide range of progressive stages that include a spectrum of liver diseases, from benign steatosis to NASH, cirrhosis and, ultimately, HCC. Even though most patients develop simple steatosis, approximately 7–30 % of them progress to NASH. Thereafter, the disease may evolve to cirrhosis within 10 years, and ultimately lead to an increased risk of developing HCC( Reference Ekstedt, Franzén and Mathiesen 27 ).
NAFLD pathogenesis is complex and involves different cellular and molecular mechanisms that promote the development of liver cancer. The progression to HCC in the context of NAFLD is a gradual process that follows a dysplasia–carcinoma sequence that may take several years to develop. During this period, erratic liver remodelling with repeated cycles of hepatocellular destruction and compensatory proliferation as a result of oxidative stress, inflammation, altered immunity and fibrosis create a propitious environment for the development of carcinogenesis( Reference Farazi and De Pinho 28 ). Throughout this process, interaction between many different oncogenic pathways and hepatic cell types occurs. As a result, hepatic stellate cells (HSC), sinusoidal endothelial cells (SEC), hepatic lymphocytes and Kupffer cells (KC) acquire pro-oncogenic properties and, at the same time, produce different growth factors, chemokines and cytokines that foster the beginning and progression of HCC( Reference Wu, Ma and Fang 29 ).
Even though it has been reported that most cases of NAFLD-related HCC develop in the context of cirrhosis, NAFLD is generally associated with obesity and diabetes, which are independently connected with an augmented risk for cancer( Reference Younossi, Gramlich and Matteoni 30 ). In addition, it is important to highlight the link between early-life malnutrition and the metabolic syndrome in adult life. Thus, the interaction between different oncogenic pathways associated with adipose tissue dysfunction, early-life malnutrition and cirrhosis can provide the basis for the development of HCC in adulthood( Reference Brumbaugh and Friedman 2 , Reference Dietrich and Hellerbrand 25 ).
Different mechanisms of oncogenesis such as structural genomic defects, epigenetic alterations that compromise different regulatory pathways, and aberrant signalling pathways contribute to a tumour-promoting environment in NAFLD, and have been involved in HCC development( Reference Park, Lee and Yu 31 ). In this respect, the chronic inflammatory response that accompanies obesity and undernutrition, which leads to the release of pro-inflammatory cytokines, is conducive to an increased risk of developing cancer( Reference Park, Lee and Yu 31 ). In particular, TNF-α has been shown to be an important cytokine released by adipose tissue, which can activate diverse pro-oncogenic pathways such as the c-Jun N-terminal kinase (JNK), the mammalian target of rapamycin (mTOR), the extracellular signal-regulated kinase (ERK) and the NF-κB( Reference Stickel and Hellerbrand 32 ). In addition, IL-6 has been demonstrated to have anti-apoptotic effects through activation of the signal transducer and activator of transcription 3 (STAT3)( Reference Park, Lee and Yu 31 ). Moreover, adipose-derived hormones, such as adiponectin and leptin, can contribute to this tumorigenic environment. While adiponectin has anti-inflammatory effects and is expressed at low levels in NAFLD, leptin levels are elevated in this pathology and exert pro-inflammatory and pro-fibrogenic effects( Reference Mirza 33 ). In addition, the ectopic deposition of fat in the liver and its associated cellular dysfunction may contribute to the development of HCC in the context of NAFLD. This process known as lipotoxicity can either promote oncogenic mechanisms( Reference Vinciguerra, Carrozzino and Peyrou 34 ) or contribute to the increase of lipid peroxides that can induce oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress and apoptosis( Reference Wei, Wang and Topczewski 35 , Reference Malhi, Bronk and Werneburg 36 ). In conjunction, these molecular events contribute to insulin resistance, which activates proliferative signalling pathways involved in HCC( Reference Gallagher and Le Roith 37 ).
Epigenetic mechanisms underlying non-alcoholic fatty liver disease related to malnutrition
Epigenetic mechanisms play an important role in the regulation of genomic functions, without affecting the primary DNA sequence. These alterations include post-translational modifications of histones, DNA methylation and microRNA (miRNA). In particular, during early development an embryo is especially vulnerable to environmental effects, which can influence these epigenetic mechanisms. Moreover, it has been demonstrated that nutrition induces epigenetic alterations that are transmitted over many generations( Reference Jang and Serra 38 ). Since these epigenetic alterations are sensitive to environmental factors, they can provide a mechanistic link between nutrition, developmental programming and disease.
Nutritional imbalances, such as overnutrition and undernutrition, induce metabolic alterations during the early developmental period of life. These imbalances lead to epigenetic dysregulation which, in turn, is associated with the development of NAFLD. Moreover, these epigenetic alterations have been proposed as important predetermining factors that affect the individual susceptibility to develop NAFLD( Reference Lee, Friso and Choi 39 ). Thus, several epigenetic alterations that contribute to pathophysiology of NAFLD are discussed below.
DNA methylation
Among epigenetic mechanisms, DNA methylation arises as a potent regulator of gene expression, whose intervention has been strongly related to carcinogenesis( Reference Franco, Schoneveld and Georgakilas 40 ). DNA methylation is a modification that occurs at cytosine nucleotides, in particular in those which precede a guanine, usually referred as CpG islands. These CpG islands are typically located in promoter regions of oncogenes, DNA repair genes or tumour-suppressor genes( Reference Lahtz and Pfeifer 41 ), thus affecting gene expression in several cancers. In this respect, it has been shown that while hypermethylation blocks gene transcription, hypomethylation induces gene activation. This degree of DNA methylation depends on the activity of DNA methyltransferases (DNMT).
Nutrition is strongly related to DNA methylation since this epigenetic mechanism depends on the availability of S-adenosylmethionine (SAM), which needs methyl donors from food (folate, choline and betaine) for its synthesis( Reference Niculescu and Zeisel 42 ). In this respect, methyl-deficient diets were reported to induce liver injury in a similar way as that observed in human NASH patients( Reference Anderson, Sant and Dolinoy 43 ). Moreover, this diet was shown to affect DNA methylation as a consequence of an important decrease in liver SAM concentration. At the same time, it resulted in the differential expression of more than a hundred genes associated with DNA damage and repair, lipid and glucose metabolism, fibrosis, and liver tissue remodelling. In this regard, the number of hypomethylated genes was significantly greater than the number of hypermethylated ones( Reference Lillycrop, Phillips and Torrens 44 ). Another study reported that in a model of intra-uterine growth restriction, using rats exposed to a low-protein diet during pregnancy, the placenta showed hypomethylation of the wingless-type MMTV integration site family member 2 (Wnt2) promoter, which is associated with fetal growth( Reference Reamon-Buettner, Buschmann and Lewin 45 ). Moreover, it was evidenced that a low-protein diet during pregnancy was conducive to DNA hypermethylation of H19/insulin-like growth factor II (IGF2) genes that resulted in up-regulation of DNMT1 and DNMT3 in the liver of male offspring( Reference Gong, Pan and Chen 46 ).
As was explained before, DNA methylation can be inherited from parents. It has been shown that exposure to a high-fat diet (HFD) during pregnancy can induce hypomethylation of cyclin-dependent kinase inhibitor 1a (CDKN1a), a liver cell cycle inhibitor, that acts during the early postnatal life of the offspring( Reference Dudley, Sloboda and Connor 47 ). Given that CDKN1a up-regulation has been related to hepatocyte growth in liver disease, this result suggests that a HFD in this period induces an early liver dysfunction, which may later predispose to NAFLD. Another study demonstrated that consumption of a Western diet during pregnancy and early postnatal life is conducive to an augmented susceptibility to NAFLD in male offspring( Reference Pruis, Lendvai and Bloks 48 ).
Liver steatosis plays a key role in NAFLD pathophysiology. It requires coordination between PPARα and PPARγ for balance between fatty acid synthesis and oxidation( Reference Varga, Czimmerer and Nagy 49 ). While PPARα has been shown to be involved in the modulation of peroxisomal and mitochondrial β-oxidation in liver and skeletal muscle, PPARγ, on the other hand, has been found to be central in the regulation of adipogenesis and to decrease the expression of proinflammatory cytokines. It has been reported that PPARα expression is down-regulated in liver steatosis, thus contributing to lipogenesis( Reference Sun, Fan and Qiao 50 ). This effect can be aggravated by the up-regulation of PPARγ, as a result of PPARγ promoter hypomethylation( Reference Giby and Ajith 51 ). In addition, the epigenetic alteration of liver PPARγ in NAFLD patients has been shown to contribute to insulin resistance. Furthermore, methylation levels of PPARγ coactivator 1α (PGC1α) promoter correlated with the homeostatic model assessment of insulin resistance (HOMA-IR) and fasting insulin, while those of mitochondrial transcription factor A (TFAM) promoter were inversely correlated with fasting insulin( Reference Sookoian, Rosselli and Gemma 52 ).
Mitochondrial DNA methylation has also been involved in the development of NAFLD( Reference Chen, Broséus and Hergalant 53 ). It has been reported that mitochondrial NADH dehydrogenase 6 (MT-ND6) is strongly methylated, which leads to MT-ND6 mRNA down-regulation in NASH patients. Moreover, hepatic methylation of MT-ND6 was shown to be associated with NAFLD( Reference Pirola, Gianotti and Burgueño 54 ).
Although there are more data from animal models of NAFLD, DNA methylation alterations have also been reported in NAFLD patients. Furthermore, nearly 70 000 methylated CpG sites in genes related to tissue repair and metabolic regulation have been found in these patients( Reference Murphy, Yang and Moylan 55 ). Another study revealed the methylation of nine genes related to intermediate metabolism and insulin-like signalling, which are strongly associated with NAFLD( Reference Ahrens, Ammerpohl and von Schönfels 56 ). In addition, it has been demonstrated that those who experienced famine in utero, as a result of the Dutch ‘Hunger Winter’, presented an altered methylation of the insulin-like growth factor-2 (IGF2) gene in adult life( Reference Heijmans, Tobi and Stein 57 ).
Histone modifications
Epigenetic alterations related to NAFLD are not restricted to DNA methylation. In fact, aberrant histone modifications have been reported to promote the development of insulin resistance, thereby playing a key role in NAFLD( Reference Ling and Groop 58 ). Histone tails can be post-transcriptionally modified by acetylation, methylation, ubiquitinylation, phosphorylation and SUMOylation, thus determining if chromatin will be active or not and, as a result, affecting the expression of genes within that portion of chromatin( Reference Jang and Serra 38 ).
Acetylation on lysine residues has been shown to be the most frequent histone modification. While acetylation is related to activation of gene transcription and is catalysed by histone acetyltransferases (HAT), deacetylation is associated with gene inactivation and is catalysed by histone deacetylases (HDAC). In fact, it has been evidenced that the imbalance between HDAC and HAT affects histone acetylation, thereby altering gene expression in NAFLD( Reference Tian, Wong and Chan 59 ). Among HDAC, sirtuins (SIRT) have been reported to be involved in multiple cellular events related to energy homeostasis. In particular, SIRT1, the most studied member of the family, has been shown to regulate metabolic processes such as fatty acid oxidation, gluconeogenesis, lipolysis and mitochondrial activity( Reference Feige and Auwerx 60 ). Given that SIRT1 inhibits NF-κB action and, in doing so, decreases the inflammatory response, its liver deletion augments steatosis and obesity-induced inflammation. On the other hand, SIRT1 overexpression exerts a beneficial effect against steatosis and insulin resistance( Reference Purushotham, Schug and Xu 61 ). The deletion of SIRT3 in mice, which is localised in mitochondria, has been associated with alterations compatible with NAFLD( Reference Hirschey, Shimazu and Jing 62 ). In addition, a HFD during pregnancy was found to induce the reduction of fetal liver HDAC1, suggesting that this diet can affect fetal chromatin through histone modifications( Reference Aagaard-Tillery, Grove and Bishop 63 ). Another study reported that the HAT activator p300, which is related to NAFLD progression, modulates carbohydrate-responsive element-binding protein (ChREBP), inducing its hyperacetylation, thus promoting its transcriptional activity. Moreover, this epigenetic modification in p300 led to liver lipogenesis in mice and was associated with insulin resistance( Reference Bricambert, Miranda and Benhamed 64 ). In addition, it has been proven that alterations in the regulation of circadian clock genes, as a result of HDAC3 action, can induce inadequate lipid metabolism in the liver, thereby helping in the development of NAFLD( Reference Feng, Liu and Sun 65 , Reference Mazzoccoli, Vinciguerra and Oben 66 ).
Histone methylation is different between cell types and has been related to fetal development. This histone modification can occur in lysine as well as in arginine residues in H3 and H4 histones, and can be reversed by histone demethylases. The histone methyltransferase PRMT1 has been shown to induce PPARγ co-activator 1α (PGC1α) methylation, thus altering fatty acid oxidation and energy metabolism( Reference Gueant, Namour and Gueant-Rodriguez 67 ). On the other hand, the histone demethylase JHDM2A has been reported as a regulator in energy homeostasis and fat deposition( Reference Portha, Fournier and Kioon 68 ).
MicroRNA
Among epigenetic mechanisms, miRNA have been strongly related to the development of NAFLD. miRNA are non-coding single-stranded RNA that modulate mRNA degradation or inhibition of translation, thus affecting the expression of different genes. It has been shown that one miRNA can alter the expression of multiple genes and, on the other hand, multiple miRNA can affect a single gene. Moreover, miRNA can modulate DNA methylation and histone modifications( Reference Lynn 69 ).
The aberrant expression of miRNA has been related to insulin resistance, obesity and NAFLD( Reference Lakner, Bonkovsky and Schrum 70 ). It has been reported that almost 100 miRNA are differentially expressed in NASH patients( Reference Cheung, Puri and Eicken 71 ). In another study, it was shown that forty-six miRNA were distinctively expressed in NASH patients, with twenty-three of them being found up-regulated, including miRNA-21, miRNA-100 and miRNA-34a, and the rest down-regulated, such as miRNA-126 and miRNA-122( Reference Cheung, Puri and Eicken 71 ). A maternal HFD during gestation and lactation has also been proven to alter the expression of twenty-three miRNA in offspring liver( Reference Zhang, Zhang and Didelot 72 ). In another study, it was shown that the differential expression of miRNA was related to the progression from liver steatosis to HCC, suggesting that each pathological state involves diverse miRNA( Reference Tessitore, Cicciarelli and Del Vecchio 73 ).
In particular, miRNA122, the principal hepatic microRNA, was found to be down-regulated in NAFLD patients compared with a control group( Reference Zhang, Zhang and Didelot 72 ). Moreover, the deletion of this liver-specific miRNA in mice induced hepatic steatosis, inflammation and HCC( Reference Hsu, Wang and Kota 74 ). As a result, this miRNA has been proposed to be a major contributor to the regulation of lipid metabolism in the liver, thus having a tumour-suppressor role in this organ( Reference Wen and Friedman 75 ).
Apart from miRN-122, there are other miRNA that have been related to NAFLD. The miRNA 146b, 143, 34a and 23a were reported to be significantly increased in NAFLD( Reference Tian, Wong and Chan 59 ). Other miRNA have been shown to modulate lipid and cholesterol regulatory genes, such as miRNA 370, 33, 103 and 104, thus ultimately contributing to the development of NAFLD.
Role of the immune response in non-alcoholic fatty liver disease progression
The liver is a site where complex immunological activity occurs, which involves different immune cells as well as non-haematopoietic cell populations. Although this organ provides a tolerogenic environment, aberrant activation of the immune system can induce harmful inflammation that firstly leads to low-level inflammation, tissue and organ injury, and ultimately fibrosis and carcinogenesis( Reference Seki and Brenner 76 ). In this respect, liver disease progression is accompanied by metabolic and inflammatory disorders that, in conjunction with genetic and environmental factors, promote a constant activation of the immune system. In particular, obesity augments the risk of NAFLD progression with a prevalence of >25 % of NASH patients among obese individuals( Reference Machado, Marques-Vidal and Cortez-Pinto 77 ). Thus, inflammatory processes in the liver are involved in both homeostasis and pathology. When adequate immune activation occurs due to pathogens or tissue damage, liver homeostasis is preserved by the resolution of inflammation. It has been demonstrated that immune defects commonly arise in under- and overnourished children. Those who suffer obesity and the metabolic syndrome display immune activation due to perturbed energy utilisation( Reference Gregor and Hotamisligil 78 ), and those with severe acute malnutrition exhibit elevated systemic proinflammatory cytokines, NEFA and ketones( Reference Bartz, Mody and Hornik 79 ). The following therapeutic feeding reduces inflammation in those children( Reference Bartz, Mody and Hornik 79 ). However, when inflammation is deregulated, pathology and organ injury arise; thus early-life malnutrition represents an important risk factor.
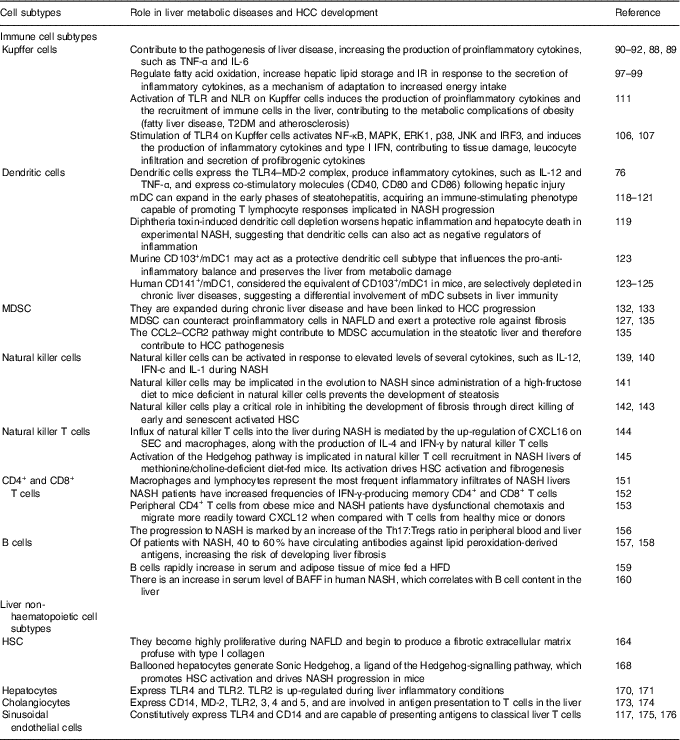
As mentioned above, the liver is comprised by both immune cell and non-haematopoietic cell populations. Liver myeloid immune cell populations are represented by KC, myeloid dendritic cells (mDC) and plasmacytoid dendritic cells (pDC), and myeloid-derived suppressor cells (MDSC)( Reference Robinson, Harmon and O’Farrelly 80 ). The innate lymphocytes in the adult liver include natural killer (NK) cells, NK T (NKT) cells, mucosal associated invariant T cells and γδ T cells. This organ also contains adaptive immune lymphoid cells, such as CD4+ and CD8+ T cells as well as B cells( Reference Robinson, Harmon and O’Farrelly 80 ). Non-haematopoietic cell populations are represented by HSC, hepatocytes, cholangiocytes and SEC( Reference Robinson, Harmon and O’Farrelly 80 ). These different cell populations overall enable the liver to fight in response to pathogens and endogen harmful signals. The role of immune and non-haematopoietic cell subtypes in liver metabolic diseases and HCC development are summarised at Table 1.
Table 1 Role of immune and non-haematopoietic cell subtypes in liver metabolic diseases and hepatocellular carcinoma (HCC) development

IR, insulin resistance; TLR, Toll-like receptor; NLR, NOD-like receptor; T2DM, type 2 diabetes mellitus; MAPK, mitogen-activated protein kinases; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; IRF3, interferon regulatory factor 3; IFN, interferon; MD-2, myeloid differentiation factor 2; mDC, myeloid dendritic cells; NASH, non-alcoholic steatohepatitis; MDSC, myeloid-derived suppressor cells; NAFLD, non-alcoholic fatty liver diseases; CCL2, C-C motif chemokine ligand 2; CCR2, C-C motif chemokine receptor; HSC, hepatic stellate cells; CXCL16, chemokine C-X-C motif ligand; SEC, sinusoidal endothelial cells; Tregs, regulatory T cells; HFD, high-fat diet; BAFF, B cell activating factor.
Kupffer cells
Liver-resident macrophages, known as KC, have an important role in the immune response. Given that they are the body’s primary line of defence against micro-organisms or endogenous signals( Reference Smedsrod, De Bleser and Braet 81 ), their location is critical for KC to carry out their different functions in liver (phagocytosis, antigen processing and presentation and secretion of different proinflammatory mediators such as cytokines, prostanoids, NO and reactive oxygen species)( Reference Bilzer, Roggel and Gerbes 82 ). KC feature with a vast array of pattern recognition receptors (PRR)( Reference Su, Klein and Aminlari 83 ), complement receptors( Reference Schieferdecker, Schlaf and Jungermann 84 ) and Fc receptors( Reference van Egmond, van Garderen and van Spriel 85 ), and are able to respond to cytokine, Toll-like receptor (TLR)( Reference Wu, Meng and Jiang 86 ), RIG-like receptor and NOD-like receptor (NLR) signalling( Reference Miura, Yang and van Rooijen 87 ). However, to maintain the steady state, KC can mount opposite responses to exogenous signals, polarising to M1 (classical) or M2 (alternative) phenotypes( Reference Sica and Mantovani 88 ). The M1 phenotype is characterised by the increment in the production of inflammatory cytokines, reactive nitrogen species and reactive oxygen species, and is related to microbicidal and tumoricidal activities. On the other hand, M2 macrophages exhibit immunomodulatory functions and are important in immunity against parasites and tissue remodelling( Reference Sica and Mantovani 88 ).
KC can promote the pathogenesis of liver disease, augmenting the production of proinflammatory cytokines( Reference Sica and Mantovani 88 , Reference Biswas and Mantovani 89 ). It has been demonstrated that TNF-α and IL-6 are closely related to steatosis, insulin resistance and inflammatory disorders( Reference Paz, Hemi and Le Roith 90 ). Furthermore, IL-6 expression correlates with the degree of hepatic inflammation, and fibrosis( Reference Abiru, Migita and Maeda 91 , Reference Haukeland, Damas and Konopski 92 ). Contrariwise, adiponectin shifts KC polarisation to the M2 anti-inflammatory phenotype( Reference Bonizzi and Karin 93 , Reference Lawrence and Gilroy 94 ), preventing NASH development( Reference Hagemann, Lawrence and McNeish 95 ). The lack of adiponectin induces hepatic steatosis progression, fibrosis and HCC( Reference Gatselis, Ntaios and Makaritsis 96 ). In addition, as a mechanism of adaptation to increased energy intake, KC can regulate fatty acid oxidation, increase hepatic lipid storage and insulin resistance( Reference Takeda, O’Dea and Doedens 97 ). These events occur in response to the secretion of inflammatory cytokines, thus suggesting a beneficial role for M2 KC in metabolic disorders( Reference Han, Jung and Morel 98 , Reference Liu, Chen and Wang 99 ). Stimulating M2 macrophages induces M1 macrophage apoptosis that preserves against NAFLD progression(100). Moreover, macrophage-mediated inflammation is associated with TLR activation. TLR signalling is related to hepatic damage, NAFLD, fibrosis and HCC( Reference Ekihiro and Brenner 101 – Reference Iimuro and Fujimoto 103 ). It has been shown that the TLR involved in NAFLD beginning and progression are TLR2, TLR4 and TLR9( Reference Miura, Kodama and Inokuchi 104 , Reference Rivera, Gaskin and Allman 105 ). Stimulation of TLR4 on KC activates NF-κB, mitogen-activated protein kinases (MAPK), extracellular signal-regulated kinase 1 (ERK1), p38, c-Jun N-terminal kinase (JNK) and interferon regulatory factor 3 (IRF3), and induces the production of inflammatory cytokines and type I IFN, hence promoting tissue damage, increase in leucocyte infiltration, and secretion of profibrogenic cytokines. In effect, it has been demonstrated that inactivation of TLR4 induces the attenuation of steatosis and NASH in several experimental models( Reference Csak, Velayudham and Hritz 106 , Reference Spruss, Kanuri and Wagnerberger 107 ). In addition, it was shown that TLR9–/– mice are protected from NASH and that this protection is consistent with a decrease in IL-1b production by KC( Reference Miura, Kodama and Inokuchi 104 ). Pathogen-associated molecular pattern molecules (PAMP) and damage-associated molecular pattern molecules (DAMP) can as well be recognised by NLR( Reference Martinon, Mayor and Tschopp 108 , Reference Pedra, Cassel and Sutterwala 109 ), whose activation induces the formation of the inflammasome( Reference Dixon, Flask and Papouchado 110 ). Commonly, activation of TLR and NLR leads to the production of proinflammatory cytokines and the recruitment of immune cells in the liver, as well as in other tissues, such as adipose tissue, muscle, hypothalamus, pancreatic islets and blood vessels. The resultant chronic inflammation promotes insulin resistance and energy imbalance and contributes to fatty liver disease, type 2 diabetes mellitus (T2DM) and atherosclerosis, all of which are metabolic complications of obesity( Reference Csak, Ganz and Pespisa 111 ). Of importance, inflammasome activation has been related to NAFLD development. Among various inflammasome complexes, the NLRP3 inflammasome is the best characterised and has been linked with this and other diseases( Reference Eun-Kyeong, Jin Kyung and Dong-Min 112 ). Altogether, these data support the notion that KC play a critical role in maintaining immune homeostasis of the liver. Thus, the balance of the products involved in inflammatory signalling pathways is crucial to determine NASH attenuation or progression.
Dendritic cells
Liver DC are a heterogeneous population of hepatic antigen-presenting cells whose main function is to induce T cell-mediated immunity. Hepatic DC populations are described as phenotypically immature and tolerogenic( Reference Doherty 113 ), although in an environment of chronic inflammation, DC are transformed to potent inducers of immune responses, becoming efficient antigen-presenting cells and a source of proinflammatory cytokines( Reference Rahman and Aloman 114 – Reference Thomson and Knolle 117 ).
DC are localised in the portal areas and are divided depending on the expression of specific markers into: plasmacytoid (PDCA-1+; pDC), and myeloid or classical (PDCA-1–; mDC). The latter are also subgrouped into DC103+/CD11b– type 1 (mDC1) and DC103–/CD11b+ type 2 (mDC2) cells( Reference Rahman and Aloman 114 , Reference Eckert, Klein and Kormek 115 ). Following hepatic injury DC express the TLR4–myeloid differentiation factor 2 (MD-2) complex, produce inflammatory cytokines such as IL-12 and TNF-α, and express co-stimulatory molecules (CD40, CD80 and CD86)( Reference Seki and Brenner 76 ). However, the role of DC in the pathogenesis of liver diseases is still unclear. DC ablation has been demonstrated to either prevent or worsen liver damage and inflammation depending on the experimental setting( Reference Rahman and Aloman 114 – Reference Heymann and Take 116 ). Previous reports have indicated that mDC can expand in the early phases of steatohepatitis, developing an immune-stimulating phenotype able to promote T lymphocyte responses( Reference Ibrahim, Nguyen and Rehman 118 , Reference Henning, Graffeo and Rehman 119 ) related to NASH progression( Reference Sutti, Jindal and Locatelli 120 , Reference Wolf, Adili and Piotrowitz 121 ). Nevertheless, diphtheria toxin-induced DC depletion aggravates liver inflammation and hepatocyte death in experimental NASH, suggesting that DC may also act as negative regulators of inflammation( Reference Henning, Graffeo and Rehman 119 ).
In this sense, Heier et al. ( Reference Heier, Meier and Julich-Haertel 122 ) investigated the role of type 1 myeloid DC (CD103+/DC mDC1) in the pathophysiology of steatosis and NASH. They used basic leucine zipper transcription factor, ATF-like-3 (Batf3)-deficient animals, which lack CD103+ DC and two dietary models to induce steatosis and NASH based on mice fed with a high-sucrose diet or a choline/methionine-deficient diet, respectively. It was shown that Batf3–/– mice that were administered both diets exhibited aggravated steatosis and lobular inflammation than similarly treated wild-type animals( Reference Heier, Meier and Julich-Haertel 122 ). Accordingly, CD103+/mDC1 deficiency increased liver infiltrating monocyte/macrophages as well as their release of inflammatory mediators. However, hepatic transaminases and fibrosis markers were not significantly affected( Reference Heier, Meier and Julich-Haertel 122 ). Moreover, the administration of bone marrow-derived CD103+/DC mDC1 to Batf3–/–-deficient mice was conducive to the diminishment of monocyte recruitment, hepatic transaminases and liver C-C motif chemokine ligand (CCL) 2 expression, without altering the level of steatosis( Reference Heier, Meier and Julich-Haertel 122 ). High-sucrose diet-fed Batf3–/– mice provided with bone marrow-derived CD103+/DC1 further significantly reduced proinflammatory monocyte influx along with serum TAG, cholesterol and NEFA. The authors thus proposed murine CD103+/mDC1 as a protective DC subtype that affects the pro-anti-inflammatory balance and preserves the liver from metabolic damage( Reference Heier, Meier and Julich-Haertel 122 ). It can be concluded that they are crucial participants in the inflammatory process involved in steatohepatitis in mice( Reference Bernsmeier and Albano 123 ). Moreover, human CD141+/mDC1, that are considered the equivalent of CD103+/mDC1 in mice( Reference Dutertre, Wang and Ginhoux 124 ), are abundant in healthy human livers( Reference Kelly, Fahey and Fletcher 125 ), but interestingly, while pDC and mDC2 expand in diseased livers, CD141+/DC are depleted in chronic liver diseases( Reference Kelly, Fahey and Fletcher 125 ), suggesting differential involvement of mDC subsets in liver immunity( Reference Bernsmeier and Albano 123 , Reference Kelly, Fahey and Fletcher 125 ).
In conclusion, it is evident that our understanding of the complex signalling network involved in the regulation of DC functions is incomplete, but overall these results suggest that DC may represent a key factor in the progression of NAFLD.
Myeloid-derived suppressor cells
MDSC are a heterogeneous population of immune cells defined by their ability to suppress T-cell activation through the production of IL-10, transforming growth factor-β (TGF-β) and arginase( Reference Gabrilovich and Nagaraj 126 ). Various cells in the liver, particularly HSC( Reference Höchst, Schildberg and Sauerborn 127 – Reference Hsieh, Chou and Yang 129 ), as well as liver-derived soluble factors, including hepatocyte growth factor and acute-phase proteins( Reference Yen, Yen and Hsu 130 ), can induce the differentiation of MDSC from myeloid cells. Although MDSC are also present in the healthy liver( Reference Chen, Akbar and Abe 131 ) they are expanded during chronic liver disease( Reference Pallett, Gill and Quaglia 132 ) and have been linked to HCC progression( Reference Schneider, Teufel and Yevsa 133 ).
Results from patients with liver disease and experimental animal models have been obtained, supporting the notion that MDSC are involved in the pathogenesis of liver inflammation and HCC( Reference Hammerich and Tacke 134 ). In addition, it has been reported that the depletion of hepatic MDSC increases the expression of fibrosis markers( Reference Roseboom, de Rooij and Painter 12 ), thus exerting a protective role. Moreover, it has been shown that MDSC can counteract proinflammatory cells in NAFLD( Reference Yao, Abe and Kawasaki 135 ).
In human and mouse models of liver cancer, MDSC are induced in the tumour environment and suppress anti-tumoral immune responses( Reference Hammerich and Tacke 134 ). In this sense, the CCL2–C-C motif chemokine receptor 2 (CCR) axis plays a pivotal role in the migration of MDSC in cancer, since the impairment of CCL2–CCR2 signalling inhibits tumour growth( Reference Huang, Lei and Zhao 136 – Reference Hale, Itani and Buchta 138 ). Yao et al. ( Reference Yao, Abe and Kawasaki 135 ) have found that the expression of CCL2 is up-regulated in the livers of NAFLD mice, and that CCL2 stimulates the migration of MDSC in vitro. Thus, the CCL2–CCR2 pathway might contribute to MDSC accumulation in the steatotic liver, therefore promoting HCC pathogenesis.
Innate and adaptive immune lymphoid cells
Hepatic innate lymphocyte populations (NK cells, NKT cells, mucosal associated invariant T cells and γδ T cells) are potent cytokine producers that influence both innate and adaptive immune responses in the liver. Some studies indicate that NK cells can be activated in response to elevated levels of several cytokines, such as IL-12, interferon-c and IL-1 during NASH( Reference Ganz and Szabo 139 , Reference Tian, Chen and Gao 140 ). It was reported that administration of a high-fructose diet to mice deficient in NK cells prevents the development of steatosis, showing that NK cells may be implicated in the evolution to NASH( Reference Bhattacharjee Kumar, Arindkar and Das 141 ). In addition, NK cells play a critical role in inhibiting the development of fibrosis through direct killing of early and senescent activated HSC( Reference Krizhanovsky, Yon and Dickins 142 , Reference Radaeva, Wang and Radaev 143 ). Furthermore, the influx of NKT cells into the liver during NASH is mediated by enhanced expression of the chemokine (C-X-C motif) ligand (CXCL) 16 on SEC and macrophages, which binds C-X-C chemokine receptor (CXCR) type 6 on the surface of NKT cells( Reference Wehr, Baeck and Heymann 144 ). In this respect, up-regulation of CXCL16 is accompanied by the production of IL-4 and IFN-γ by NKT cells, which worsens inflammation via macrophage activation( Reference Wehr, Baeck and Heymann 144 ). Activation of the Hedgehog pathway has also been implicated in NKT cell recruitment in NASH livers of methionine/choline-deficient diet-fed mice. Moreover, activation of the Hedgehog pathway drives HSC activation and fibrogenesis( Reference Syn, Agboola and Swiderska 145 ). Then, NKT cells seem to be involved in the development of NASH.
On the other hand, populations of adaptive lymphocytes (CD4+ and CD8+ T cells and B cells) are found in healthy livers( Reference Norris, Collins and Doherty 146 , Reference Pruvot, Navarro and Janin 147 ). Depending on the cytokine environment, T CD4+ cells can assume a proinflammatory phenotype (Th1) or an anti-inflammatory phenotype (Th2)( Reference Romagnani 148 ). The balance between Th1 and Th2 T cells is important to maintain immune system homeostasis. There is a third type of CD4+ cells called Th17, which play a pro-inflammatory role( Reference Tang, Bian and Zhao 149 ). The implication of the adaptive immunity in stimulating adipose tissue inflammation has been well studied in obesity( Reference Sell, Habich and Eckel 150 ). A similar mechanism is implicated in the beginning of inflammation in NASH, where different studies have demonstrated that both macrophages and lymphocytes are the most common inflammatory infiltrates in these livers( Reference Brunt 151 ). For instance, individuals with NASH have augmented frequencies of IFN-γ-producing memory CD4+ and CD8+ T cells( Reference Inzaugarat, Ferreyra Solari and Billordo 152 ). Dysfunctional chemotaxis is one of the molecular mechanisms driving T cell infiltration into the liver. In this sense, it has been shown that peripheral CD4+ T cells from both obese mice and NASH patients migrate more promptly toward the chemokine CXCL12 when compared with T cells from healthy animals or donors( Reference Boujedidi, Robert and Bignon 153 ). Likewise, Th17 cell infiltration is found in NASH livers and it is well known that IL-17 secretion exacerbates hepatic steatosis and inflammation( Reference Tang, Bian and Zhao 149 ). Moreover, IL-17 signalling in HSC up-regulates the expression of profibrotic genes while absence of IL-17 in a murine model of liver fibrosis diminishes the levels of proinflamamtory cytokines and cell death( Reference Tan, Qian and Jiang 154 , Reference Meng, Wang and Aoyama 155 ). In addition, different studies have reported that the progression to NASH is marked by an increment of the Th17:regulatory T cells (Tregs) ratio in peripheral blood and liver( Reference Rau, Schilling and Meertens 156 ).
B cells play an important role in NASH pathogenesis. It has been shown that 40–60 % of patients with NASH have circulating antibodies against lipid peroxidation-derived antigens( Reference Albano, Mottaran and Vidali 157 ). In addition, the elevated titres of these antibodies are in parallel with an increased risk of developing liver fibrosis( Reference Nobili, Parola and Alisi 158 ). Furthermore, Winer et al. ( Reference Winer, Winer and Shen 159 ) have demonstrated that B cells rapidly increase in serum and adipose tissue of mice fed a HFD.
B cell activating factor (BAFF) is a well-known cell survival and maturation factor for B cells. An increase in serum level of BAFF in human NASH has been demonstrated, and the serum BAFF level correlates with B cell content in the liver( Reference Kawasaki, Abe and Tada 160 ). Moreover, BAFF receptor-deficient mice showed improved obesity and insulin resistance induced by HFD but, in addition, displayed enhanced hepatic steatosis that indicates a protective role of BAFF in hepatic steatosis( Reference Kawasaki, Abe and Tada 160 , Reference Kim and Do 161 ). Nevertheless, to better understand the B cell’s contribution in sustaining hepatic inflammation and promoting fibrosis in NASH, further studies will be required.
Hepatic non-haematopoietic cells
In addition to hepatic immune cell populations, non-haematopoietic cells in the liver play key roles in local and systemic innate immunity and inflammation. HSC reside in the space between hepatocytes and the liver SEC, known as the space of Disse( Reference Weiskirchen and Tacke 162 ). In physiological conditions, HSC store vitamin A and lipids( Reference Weiskirchen and Tacke 162 , Reference Thapa, Chinnadurai and Velazquez 163 ). However, in response to a liver injury, such as in NAFLD, HSC become activated, transdifferentiate to myofibroblasts and begin to synthesise large amounts of extracellular matrix, leading to fibrosis( Reference Ramadori and Saile 164 – Reference Dranoff, Kruglov and Robson 166 ). HSC activation is a result of the release of proliferative and fibrogenic cytokines, in particular platelet-derived growth factor (PDGF) and TGF-β from different cells, such as hepatocytes, SEC, macrophages, NK cells and lymphocytes( Reference Elpek 167 ). Guy et al. ( Reference Guy, Suzuki and Zdanowicz 168 ) have found that ballooned hepatocytes generate Sonic Hedgehog, a ligand of the Hedgehog-signalling pathway that induces HSC activation and contributes to NASH progression in mice. TLR4 is another factor implicated in the activation of HSC( Reference Seki and Brenner 76 , Reference Seki, De Minicis and Osterreicher 169 ). Hepatocytes express TLR4 and TLR2, and it has been shown that TLR2 is up-regulated during inflammatory conditions( Reference Liu, Gallo and Green 170 , Reference Matsumura, Degawa and Takii 171 ). In addition, a role for biliary epithelial cells in portal and septal fibrosis has been demonstrated( Reference Chiba, Sasaki and Kitamura 172 ). Murine biliary cells express CD14, myeloid differentiation factor 2 (MD-2), TLR2, TLR3, TLR4 and TLR5( Reference Harada, Ohira and Isse 173 ), and are involved in antigen presentation to T cells( Reference Lleo and Invernizzi 174 ), thus playing an additional role in hepatic immune function. In addition, SEC constitutively express TLR4 and CD14( Reference Uhrig, Banafsche and Kremer 175 ), and are capable of presenting antigens to classical T cells( Reference Thomson and Knolle 117 , Reference Crispe 176 ).
Epigenetic changes affecting immune functions in the context of non-alcoholic fatty liver disease and hepatocellular carcinoma
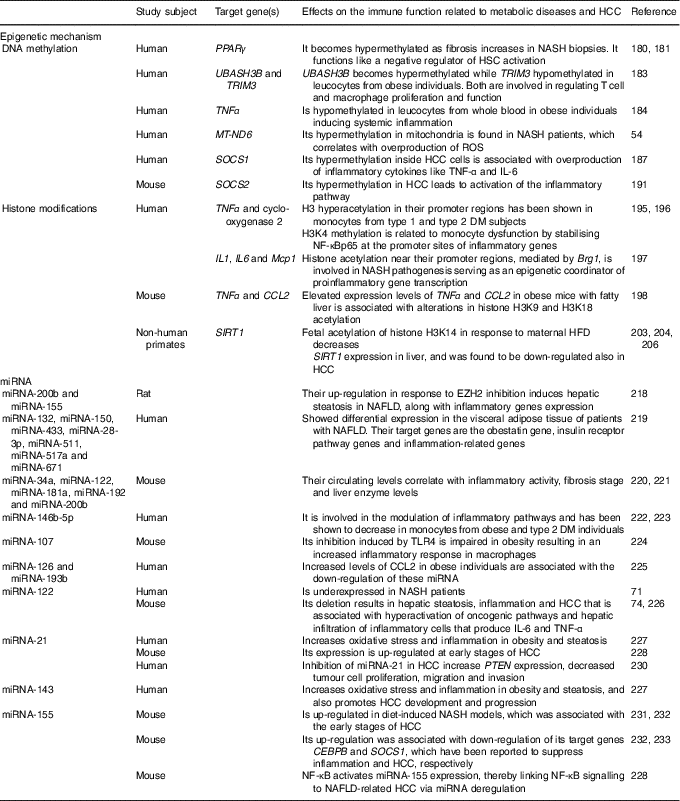
Perturbations in the immune system are characteristic of metabolic diseases and cancer development( Reference Tian, Wong and Chan 59 ). Epigenetic mechanisms are involved in the stimulation and maintenance of immune activation and inflammation. Environmental factors such as nutrition can induce epigenetic modifications( Reference Barrès, Kirchner and Rasmussen 177 ). The expression of infant immune and metabolic genes is regulated by epigenetic modifications inherited by malnourished parents( Reference Donkin, Versteyhe and Ingerslev 178 , Reference Martínez, Pentinat and Ribó 179 ). Moreover, strong evidence indicates that pre-existing epigenetic marks within genes respond to environmental stimuli by activating or repressing gene expression. In this sense, male mice that received protein–energy malnutrition during gestation showed different epigenetic marks at the liver X receptor alpha (LXR-α) locus that were inherited by their appropriately fed offspring( Reference Martínez, Pentinat and Ribó 179 ). The LXR-α gene is involved in inflammation and lipid metabolism, and LXR-α-dependent modifications in liver lipid-synthesis genes were clearly visible in the second generation( Reference Martínez, Pentinat and Ribó 179 ). Epigenetic changes, hence, would play a primordial role in the interplay between genetics and environmental factors to drive the inflammatory changes accompanying diet-induced metabolic diseases that could even end in cancer( Reference Tian, Wong and Chan 59 ). DNA methylation, covalent modification of histones and the expression of microRNA are the epigenetic phenomena involved in inflammatory processes. The epigenetic mechanisms for NAFLD and HCC development are summarised at Table 2.
Table 2 Epigenetic mechanisms affecting the immune response involved in liver metabolic diseases and hepatocellular carcinoma (HCC)

NASH, non-alcoholic steatohepatitis; HSC, hepatic stellate cells; UBASH3B, ubiquitin-associated and SH3 domain-containing protein B; TRIM3, tripartite motif-containing 3; MT-ND6, mitochondrial NADH dehydrogenase 6; ROS, reactive oxygen species; SOCS, suppressor of cytokine signalling; DM, diabetes mellitus; H, histone; K, lysine; NF-κBp65, NF-κB p65 subunit; Mcp1, monocyte chemoattractant protein-1; Brg1, Brahma-related gene; CCL2, C-C motif chemokine ligand 2; SIRT1, sirtuin 1; HFD, high-fat diet; miRNA, microRNA; EZH2, zeste homolog 2; NAFLD, non-alcoholic fatty liver diseases; TLR4, Toll-like receptor 4; PTEN, phosphatase and tensin homolog gene; CEBPB, CCAAT/enhancer binding protein β.
DNA methylation
DNA methylation is one of the main epigenetic modifications that contribute to altered gene expression in NAFLD. As mentioned above, PPARγ has been found to reduce the expression of proinflammatory cytokines and contribute to insulin resistance in patients with NAFLD( Reference Sun, Fan and Qiao 50 – Reference Sookoian, Rosselli and Gemma 52 ). It has been shown that the promoter region of PPARγ becomes hypermethylated as fibrosis augments in NASH liver biopsies from patients. Thus, it functions like a negative regulator of HSC activation and liver fibrogenesis( Reference Mann, Chu and Maxwell 180 , Reference Zeybel, Hardy and Wong 181 ). Increasing evidence demonstrated that DNA methylation and insulin resistance in NAFLD patients are critical players for the transition from steatosis to fibrotic NASH( Reference Tian, Wong and Chan 59 ).
It is well known that NAFLD is linked to obesity and the metabolic syndrome( Reference Younossi, Stepanova and Afendy 182 ). Adipose tissue enlargement related to the metabolic syndrome is associated with immune system activation and chronic inflammation, which is also involved in the development of insulin resistance( Reference Sell, Habich and Eckel 150 ). In addition, altered methylation levels on the UBASH3B (ubiquitin-associated and SH3 domain-containing protein B) and TRIM3 (tripartite motif-containing 3) genes in obesity have been reported, genes which are both involved in regulating T cell and macrophage proliferation and function( Reference Wang, Zhu and Snieder 183 ). Another study has succeeded in linking altered DNA methylation to systemic inflammation in obesity. It was shown that TNF-α promoter was hypomethylated in leucocytes from whole blood in obese individuals( Reference Hermsdorff, Mansego and Campión 184 ). Moreover, another study identified global DNA hypermethylation in B cells from obese and T2DM individuals( Reference Simar, Versteyhe and Donkin 185 ), suggesting altered B cell function in metabolic diseases. In addition, the up-regulation of the DNA DNMT3B in macrophages has been linked to elevated levels of SFA, leading to M1 polarisation and adipose tissue inflammation( Reference Yang, Wang and Liu 186 ). Furthermore, oxidative stress has certainly been linked to inflammation, and mitochondria are the principal sources and targets of reactive oxygen species. As mentioned above, elevated levels of MT-ND6 methylation in mitochondria have been found in NASH patients( Reference Pirola, Gianotti and Burgueño 54 ). When NAFLD progresses to HCC, inflammation and oxidative stress also play a critical role in this process. The suppressor of cytokine signalling (SOCS) 1 is a negative regulator of cytokine signalling, epigenetically regulated and, indeed, a suppressor of inflammation. SOCS1 promoter hypermethylation is one of the best-known epigenetic changes in macrophages and HCC( Reference Herath, Leggett and MacDonald 187 – Reference Amodio, Bellizzi and Leotta 189 ). It was demonstrated that loss of SOCS1 expression inside tumour cells through promoter hypermethylation is markedly associated with overproduction of inflammatory cytokines such as TNF-α and IL-6( Reference Cheng, Huang and Ma 190 ). Moreover, increased promoter methylation of the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) inhibitor SOCS2 also in the tumours of glycine N-methyltransferase (GNMT) knockout mice has been shown, leading to epigenetic activation of the inflammatory pathway( Reference Martinez-Chantar, Vazquez-Chantada and Ariz 191 ). Is evident that the remodelling of the immune system in metabolic disease and HCC is intimately linked to epigenetic modifications and methylation of CpG dinucleotides is the most well-studied epigenetic mechanism that plays crucial roles in cell fate decisions and oncogenic transformation till now.
Histone modifications
Modification of histones is another key epigenetic change that can alter the immune response( Reference Schoenborn, Dorschner and Sekimata 192 , Reference Di Spirito and Shen 193 ). In obese individuals, HDAC4 has been reported to be decreased in peripheral blood myeloid cells and to be also inversely correlated with the expression of the proinflammatory CCL5( Reference Abu-Farha, Tiss and Abubaker 194 ). If its expression is restituted, the activation of TNF-α-induced NF-κB is inhibited, indicating its regulatory role in inflammation. Increased histone H3 hyperacetylation in the TNF-α and cyclo-oxygenase 2 promoter regions has been shown in monocytes from type 1 diabetes mellitus and T2DM subjects( Reference Miao, Gonzalo and Lanting 195 ), while histone H3 lysine 4 (H3K4) methylation has been related to monocyte dysfunction by stabilising NF-κBp65 at the promoter sites of inflammatory genes( Reference Li, Reddy and Miao 196 ). The liver is a metabolically active organ that is usually affected by inflammation in obese individuals. In this regard, it has been demonstrated that when hepatocytes are cultured with NEFA the up-regulation of the Brahma-related gene (Brg1) and Brahma protein are induced, which promote inflammation by increasing histone acetylation near the promoter regions of the IL-1, IL-6 and monocyte chemoattractant protein-1 (Mcp1) genes. Thus, Brg1 is involved in NASH pathogenesis, behaving as an epigenetic coordinator of proinflammatory gene transcription( Reference Tian, Xu and Fang 197 ). Moreover, TNF-α and chemokine CCL2 are considered important inflammatory mediators in the development of NAFLD. By a chromatin immunoprecipitation assay, an increase in H3K9 and H3K18 acetylation at TNF-α and CCL2 in obese mice was demonstrated ( Reference Mikula, Majewska and Ledwon 198 ). These results indicate that elevated levels of TNF-α and CCL2 expression in fatty liver are associated with the alterations in histone H3 acetylation( Reference Mikula, Majewska and Ledwon 198 ). Likewise, SIRT1, a class III HDAC, which is involved in the regulation of glucose homeostasis, antihyperlipidaemic activity, insulin sensitivity, oxidative stress, anti-inflammatory activity, and antiaging activity( Reference Colak, Yesil and Mutlu 199 ), has been shown to be down-regulated in the adipose tissue of obese individuals. This results in augmented macrophage recruitment through increased chemoattractant and cytokine production( Reference Gillum, Kotas and Erion 200 ). As mentioned above, this HDAC also inhibits NF-κB activity to diminish the inflammatory response and modulates other cytokines implicated in lipid metabolism( Reference Tian, Wong and Chan 59 , Reference Herranz and Serrano 201 ). Therefore, liver-specific deletion of SIRT1 has been shown to enhance fatty liver disease and obesity-induced inflammation, while SIRT1 overexpression exhibited a protective effect against steatohepatitis and insulin resistance( Reference Purushotham, Schug and Xu 61 , Reference Escande, Chini and Nin 202 ). In addition, another study performed in non-human primates has demonstrated that a maternal HFD increases the fetal acetylation of histone H3K14 and decreased SIRT1 expression in their fetal livers( Reference Suter, Chen and Burdine 203 ). Moreover, a significant reduction in the expression of SIRT1 in NAFLD animal models has been reported, and natural SIRT1 activator has shown protective effects on metabolic diseases( Reference Colak, Ozturk and Senates 204 ). Furthermore, the mitochondrial SIRT3 has been shown to be required for maintenance of integrity in response to oxidative stress( Reference Kim, Patel and Muldoon-Jacobs 205 ). Mice deficient in SIRT3 displayed NASH( Reference Hirschey, Shimazu and Jing 62 ), therefore indicating its role in regulating inflammation. Otherwise, several studies have demonstrated that aberrant chromatin modifications link metabolic perturbation to cellular transformation in the liver. In this sense, the expression of SIRT1 was found to be down-regulated in HCC, as it happens in NAFLD( Reference Wang, Sengupta and Li 206 ). Indeed, overexpression of SIRT1 in a HFD model exposed to hepatic carcinogen protected mice from HCC( Reference Herranz, Muñoz-Martin and Canamero 207 ), indicating a tumour-suppressive role for SIRT1.
Moreover, mice with genetic ablation of the enzyme that converts methionine to SAM (methionine adenosyl transferase 1A; MAT1A) displayed lower hepatic SAM levels, higher lipogenesis and oxidative stress; thus, they were predisposed to NAFLD-associated HCC( Reference Lu, Alvarez and Huang 208 , Reference Martinez-Chantar, Corrales and Martinez-Cruz 209 ). On the other hand, mice defective in glycine N-methyltransferase, the SAM catabolic enzyme, displayed higher hepatic SAM levels but also developed NAFLD and HCC( Reference Martinez-Chantar, Vazquez-Chantada and Ariz 191 , Reference Liao, Liu and Lee 210 ). Thus, either an excess or a defect in SAM liver levels may result in aberrant methylation of histones, deriving from epigenetic modulation of critical metabolic and carcinogenic pathways( Reference Lu and Mato 211 ).
Non-coding RNA
Short non-coding miRNA and long non-coding RNA (lncRNA) act in modulating translation and transcription of target genes, thus regulating a variety of biological functions( Reference Wang, Yao and Lin 212 ). miRNA play an important role in lipid metabolism and inflammation and some of these miRNA have been shown to be epigenetically regulated in NAFLD( Reference Finch, Marquardt and Yeoh 213 – Reference Panera, Gnani and Crudele 215 ). In this sense, the enhancer of zeste homolog 2 (EZH2) is known as a histone–lysine N-methyltransferase enzyme that intervenes in histone methylation and transcriptional repression( Reference Viré, Brenner and Deplus 216 ), catalysing the addition of methyl groups to histone H3 at lysine 27( Reference Cao, Wang and Wang 217 ). Moreover, a study performed in rats demonstrated that EZH2 inhibition induced up-regulation of TNF-α and TGF-β inflammatory genes as well as of miRNA-200b and miRNA-155, subsequently inducing hepatic steatosis in NAFLD( Reference Vella, Gnani and Crudele 218 ). Another study developed in patients with NAFLD showed differential expression of seven miRNA (miRNA-132, miRNA-150, miRNA-433, miRNA-28-3p, miRNA-511, miRNA-517a and miRNA-671) in the visceral adipose tissue, which target genes comprising the obestatin gene, insulin receptor pathway genes and inflammation-related genes( Reference Estep, Armistead and Hossain 219 ).
Even though liver biopsy is the ‘gold standard’ for diagnosis of NAFLD, it is very invasive. Thus, serum miRNA panelling is currently being considered as a non-invasive technique that may be used as a biomarker for diagnosis and monitoring of NAFLD. A study showed that the levels of circulating miRNA-34a, miRNA-122, miRNA-181a, miRNA-192 and miRNA-200b were significantly correlated with inflammatory activity, fibrosis stage and liver enzyme levels in mice( Reference Cermelli, Ruggieri and Marrero 220 , Reference Tryndyak, Latendresse and Montgomery 221 ).
Different miRNA are differentially expressed both at the tissue and systemic levels in obese or T2DM individuals. The miRNA-146b-5p is involved in the modulation of inflammatory pathways and has been shown to be decreased in monocytes from obese( Reference Hulsmans, Van Dooren and Mathieu 222 ) and T2DM( Reference Balasubramanyam, Aravind and Gokulakrishnan 223 ) individuals. In addition, TLR4-induced inhibition of miRNA-107 is impaired in obesity, leading to an increased inflammatory response in macrophages( Reference Foley and O’Neill 224 ). Furthermore, the trafficking of inflammatory immune cells to the adipose tissue in obesity is controlled by the expression of chemokines( Reference Sell, Habich and Eckel 150 ). An increased level of CCL2 in obese individuals which was associated with reduced miRNA-126 and miRNA-193b has been shown( Reference Arner, Mejhert and Kulyté 225 ).
Interestingly, the liver-specific miRNA-122 is underexpressed in NASH patients( Reference Cheung, Puri and Eicken 71 ). As mentioned above, different studies have demonstrated that deletion of miRNA-122 in mice resulted in hepatic steatosis, inflammation and HCC( Reference Hsu, Wang and Kota 74 , Reference Tsai, Hsu and Hsu 226 ). These pathological manifestations were associated with hyperactivation of oncogenic pathways( Reference Hsu, Wang and Kota 74 , Reference Tsai, Hsu and Hsu 226 ) and hepatic infiltration of inflammatory cells that produced IL-6 and TNF-α( Reference Hsu, Wang and Kota 74 ). Furthermore, there is evidence indicating that miRNA-21, miRNA-103, miRNA-143 and miRNA-378 increase oxidative stress and inflammation in animal models with obesity and steatosis( Reference Hulsmans, de Keyzer and Holvoet 227 ). Using a NASH-related HCC model of rodents fed on a choline-deficient, low-methionine and amino acid-defined diet, authors found a significant increase in miRNA-21 expression at early stages of HCC( Reference Wang, Majumder and Nuovo 228 ). Moreover, unsaturated fatty acids in hepatocytes induce steatosis mediated by mammalian target of rapamycin (mTOR)/NF-κB signalling pathway stimulation and concomitantly activate miRNA-21, that in turn suppresses the expression of the phosphatase and tensin homolog gene (PTEN)( Reference Vinciguerra, Sgroi and Veyrat-Durebex 229 ). In HCC, inhibition of miRNA-21 increased PTEN expression, decreased tumour cell proliferation, migration, and invasion, suggesting that miRNA-21 and the tumour-suppressor PTEN pathway are involved in the NAFLD-related HCC development( Reference Meng, Henson and Wehbe-Janek 230 ). In addition, miRNA-143 was shown to not only regulate obesity-associated pathways but also promote HCC development and progression( Reference Tian, Wong and Chan 59 ). Furthermore, using diet-induced NASH models it was demonstrated that miRNA-155 is overexpressed in these livers and it was associated with the early stages of HCC( Reference Pogribny, Starlard-Davenport and Tryndyak 231 , Reference Yan, Jia and Chen 232 ). At these early stages, up-regulation of miRNA-155 was related to the down-regulation of their target genes CCAAT/enhancer binding protein β (CEBPB) and SOCS1 that have been shown to suppress inflammation and HCC, respectively( Reference Yan, Jia and Chen 232 , Reference Worm, Stenvang and Petri 233 ). In addition, NF-κB activates miRNA-155 expression, thus linking NF-κB signalling to NAFLD-related HCC via miRNA deregulation( Reference Wang, Majumder and Nuovo 228 ).
On the other hand, the contribution of lncRNA to obesity-related inflammation is not completely understood. lncRNA E33 has been reported to be up-regulated in macrophages from diabetic and diet-induced diabetic mice( Reference Reddy, Chen and Park 234 ). lncRNA E33 overexpression in RAW cells was associated with the up-regulation of several proinflammatory genes while lncRNA E33 silencing reversed these effects( Reference Reddy, Chen and Park 234 ). The expression of non-coding RNA, in particular miRNA, constitutes an important epigenetic mechanism that coordinates downstream features of the immune system, including immune cell differentiation, function and recruitment.
Conclusion
Early-life malnutrition induces epigenetic alterations that modify the expression of genes involved in lipid and glucose metabolism, and also in the immune response developed in the context of NAFLD. There is limited knowledge about the interaction between early-life malnutrition and its long-term effects over the epigenetic modulation of the immune function, which ultimately favours NAFLD. Therefore, the strategy of modulating the epigenome to prevent and/or treat NAFLD, cirrhosis and HCC is still challenging. To progress in this field, it is necessary to have a better understanding of the epigenetic mechanisms that alter the immune system in malnutrition. This knowledge will offer us novel treatment interventions for vulnerable individuals, which is crucial to reduce worldwide mortality.
Acknowledgements
The present review received no specific grant from any funding agency, commercial or not-for-profit sectors.
S. C., A. L. C. and A. N. C. planned the manuscript. S. C. and A. L. C. wrote the manuscript. A. N. C. provided a critical review of the paper. All authors contributed and agreed on the final version of the manuscript.
The authors have no declarations of interest to declare.