



Introduction
Non-human primates (NHPs) are among the most endangered of mammalian taxa (Estrada et al., Reference Estrada, Garber, Rylands, Roos, Fernandez-Duque, Di Fiore, Anne-Isola Nekaris, Nijman, Heymann, Lambert, Rovero, Barelli, Setchell, Gillespie, Mittermeier, Arregoitia, de Guinea, Gouveia, Dobrovolski, Shanee, Shanee, Boyle, Fuentes, MacKinnon, Amato, Meyer, Wich, Sussman, Pan, Kone and Li2017; Fernández et al., Reference Fernández, Kerhoas, Dempsey, Billany, McCabe and Argirova2022). Asian NHPs are particularly threatened, with one of the largest proportions of threatened NHP species reported across global regions, second only to Madagascar (Fernández et al., Reference Fernández, Kerhoas, Dempsey, Billany, McCabe and Argirova2022). Asian NHPs commonly feature among the International Union for Conservation of Nature's ‘World's 25 Most Endangered Primates’ and face a variety of conservation challenges (Mittermeier et al., Reference Mittermeier, Valladares-Pádua, Rylands, Eudey, Butynski, Ganzhorn, Kormos, Aguiar and Walker2006). Consequently, effective conservation strategies rely on understanding both their ecology and conservation threats. One such threat is the risk of disease, with various bacterial, viral and parasitic infections previously reported to have damaging consequences for NHP hosts (Chapman et al., Reference Chapman, Gillespie and Goldberg2005; Mul et al., Reference Mul, Paembonan, Singleton, Wich and Van Bolhuis2007; Gillespie et al., Reference Gillespie, Nunn and Leendertz2008; Bicca-Marques et al., Reference Bicca-Marques, Rabelo, de Almeida and Sales2022). In particular, host–parasite interactions have long been associated with population dynamics of wildlife hosts (Hudson and Dobson, Reference Hudson and Dobson1995). Wild NHPs host numerous parasites (Nunn et al., Reference Nunn, Altizer, Jones and Sechrest2003; Frias and MacIntosh, Reference Frias, MacIntosh, Jones-Engel and Knauf2020), including strongylid nematodes, a major radiation of parasitic helminths, herein referred to as strongylids. Strongylids are amongst the most common gastrointestinal parasites of wild NHPs, typically occurring as asymptomatic infections within such hosts (Cantacessi et al., Reference Cantacessi, Campbell and Gasser2012; Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018). However, clinical manifestations have been previously reported in both captive and wild NHPs (Pit et al., Reference Pit, Polderman, Baeta, Schulz-Key and Soboslay2001; Mul et al., Reference Mul, Paembonan, Singleton, Wich and Van Bolhuis2007; Muhangi et al., Reference Muhangi, Gardiner, Ojok, Cranfield, Gilardi, Mudakikwa and Lowenstine2021). Strongylids are expected to occur in complex communities of multiple genera within NHP hosts, as is conventional in large terrestrial herbivores (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018).
While strongylids have been widely studied in both human medicine and veterinary sciences (Zajac, Reference Zajac2006; Cantacessi et al., Reference Cantacessi, Campbell and Gasser2012), knowledge of these infections in wildlife is still limited (Krief et al., Reference Krief, Jamart, Mahé, Leendertz, Mätz-Rensing, Crespeau, Bain and Guillot2008; Mclean et al., Reference Mclean, Kinsella, Chiyo, Obanda, Moss and Archie2012; Walker and Morgan, Reference Walker and Morgan2014). It is restricted by the indistinguishability of different species, or even genera, through traditional coproscopic approaches (Jex et al., Reference Jex, Lim, Bethony, Hotez, Young and Gasser2011; Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018). Previously applied molecular approaches are time consuming and require either single larval specimens or species-specific primers. Such methodologies are insufficient for capturing the true genetic diversity of these complex communities, at risk of overlooking rarer taxa (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018). Technological advancements in high-throughput sequencing (HTS) now facilitate simultaneous sequencing of mixed DNA from entire communities, including identification of rare taxa (von Bubnoff, Reference von Bubnoff2008), in a timely and cost-effective manner. In recent research, HTS methodologies have confirmed the presence of complex strongylid communities in various African NHPs (Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019; Mason et al., Reference Mason, Petrzelkova, Kreisinger, Bohm, Cervena, Fairet, Fuh, Gomez, Knauf, Maloueki, Modry, Shirley, Tagg, Wangue and Pafco2022). However, application is not currently widespread and these methods are yet to be applied to strongylid communities of Asian NHPs.
Recent research has revealed striking differences in strongylid communities of African great apes (Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019; Ilík et al., Reference Ilík, Kreisinger, Modrý, Schwarz, Tagg, Mbohli, Nkombou, Petrželková and Pafčo2023), raising questions as to the drivers of these complex communities. Parasite phylogenies are sometimes thought to emulate the host phylogenies within which they evolved, as predicted by the host-parasite co-speciation hypothesis (Brooks, Reference Brooks1979). However, environmental parameters, such as temperature, humidity or vegetation, are known to influence longevity and transmission of parasite populations (Silangwa and Todd, Reference Silangwa and Todd1964; Callinan and Westcott, Reference Callinan and Westcott1986; Jex et al., Reference Jex, Lim, Bethony, Hotez, Young and Gasser2011). As soil-transmitted helminths, strongylids may be particularly influenced by environmental variation, due to development of infectious larvae involving free-living larval stages within the external environment (Kalousová et al., Reference Kalousová, Piel, Pomajbíková, Modrý, Stewart and Petrželková2014; Knapp-Lawitzke et al., Reference Knapp-Lawitzke, Küchenmeister, Küchenmeister, von Samson-Himmelstjerna and Demeler2014). Similarly, parasite populations are also influenced by the presence or absence of sympatric species, be that other wildlife or domestic animals (Hatcher et al., Reference Hatcher, Dick and Dunn2012). Existence of multiple potential hosts within a given area facilitates parasite sharing between host species, particularly among phylogenetically related hosts (Dallas et al., Reference Dallas, Han, Nunn, Park, Stephens and Drake2019), including NHPs (Cooper et al., Reference Cooper, Griffin, Franz, Omotayo and Nunn2012; Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019). With a direct lifecycle, strongylids rely on overlapping host ranges for transmission (Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019), without an intermediate or transport host to facilitate wider transmission.
Coproscopic studies have confirmed a high prevalence of strongylids in both South-East and East Asian primates (Arizono et al., Reference Arizono, Yamada, Tegoshi and Onishi2012; Frias et al., Reference Frias, Hasegawa, Chua, Sipangkui, Stark, Salgado-Lynn, Goossens, Keuk, Okamoto and MacIntosh2021). To date, genetic research of strongylids in Asian NHPs has typically focused on Oesophagostomum spp., the so-called ‘nodular worms’. Only one species of nodular worm, O. aculeatum, has been genetically identified in Asian NHPs thus far (Arizono et al., Reference Arizono, Yamada, Tegoshi and Onishi2012; Frias et al., Reference Frias, Stark, Salgado Lynn, Nathan, Goossens, Okamoto and MacIntosh2019; Yalcindag et al., Reference Yalcindag, Stuart, Hasegawa, Streit, Doležalová, Morrogh-Bernard, Cheyne, Nurcahyo and Foitová2021). There are few reports of other strongylid taxa in Asian NHPs. For example, a previous molecular characterization identified the presence of a single Ternidens deminutus larva in a sampled community of Bornean NHPs (Frias et al., Reference Frias, Stark, Salgado Lynn, Nathan, Goossens, Okamoto and MacIntosh2019). However, due to this previous research using single larval specimens and relying on taxa-specific primers, the strongylid communities identified cannot be deemed conclusive. To build upon current knowledge, a holistic approach better able to capture the entire strongylid community, including rare taxa, is needed.
Here, a previously optimized HTS approach (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018) is utilized to shed light on the genetic diversity of strongylid communities infecting South-East and East Asian NHPs. This approach is applied to 5 distinct NHP populations, including a total of 6 primate species. In describing the strongylid communities of Bornean orangutans (Pongo pygmaeus), one of the only Asian great apes, and several sympatric NHP species, the aim is to provide a unique insight into the host–parasite co-speciation hypothesis within great apes. Insight into within species variation of NHP strongylid communities is also provided, through comparison of Japanese macaques (Macaca fuscata) from 3 isolated localities. We hypothesize that strongylid nematodes occur in mixed communities of multiple taxa, dominated by Oesophagostomum, matching previous findings based on single-specimen genetics.
Material and methods
Sample collection
In this study, 90 individual faecal samples were utilized, non-invasively collected from 6 different wild NHP species in South-East and East Asia, precisely across 5 localities in Malaysian Borneo and Japan. In Borneo, sampling was conducted at 2 localities: the Lower Kinabatangan Wildlife Sanctuary (LKWS) and Danum Valley Conservation Area (DVCA), both of which occur in the state of Sabah. At LKWS, 36 samples representing 5 primate species were collected: crab-eating macaque (Macaca fascicularis, n = 17), southern pig-tailed macaque (Macaca nemestrina, n = 2), proboscis monkey (Nasalis larvatus, n = 4), silvery lutung (Trachypithecus cristatus, n = 5), and Bornean orangutan (Pongo pygmaeus, n = 2). Except for Bornean orangutans, which were either followed or sampled opportunistically during their visits to the Centre, LKWS samples were collected during systematic boat surveys along the Kinabatangan River, with NHP species being opportunistically sampled when encountered at sleeping sites early in the morning. Visual appearance of faeces based on softness and presence of invertebrates was used to ensure that only fresh samples were collected. Finally, 12 identified Bornean orangutans inhabiting DVCA were sampled during standard focal follows of habituated individuals. Further sampling was conducted in Japan, with 48 samples collected from the 2 subspecies of Japanese macaques (Macaca fuscata fuscata and M. f. yakui) inhabiting 3 localities: Jigokudani (n = 15), Koshima (n = 21) and Yakushima (n = 12). At these 3 localities, the primates are habituated to human presence and each sample can be attributed to a known individual. Samples from all localities were fixed in ethanol as soon as possible after collection, always on the same day, with fixed samples then stored at environmental temperature before transportation to Wildlife Research Center, Kyoto University, Japan.
DNA isolation and sequencing
Total genomic DNA was extracted from faecal samples using a QIAamp DNA stool mini kit (Qiagen, Japan) following the manufacturer's recommendations. To ensure correct host identification of samples originating from unidentified hosts in LKWS, cytochrome b (cytb) gene fragments were amplified using the primer pair L14724/H15915 (CGAAGCTTGATATGAAAAACCATCGTTG/AACTGCAGTCATCTCCGGTTTACAAGAC – respectively) (Irwin et al., Reference Irwin, Kocher and Wilson1991). Amplicons were then purified using the Agencourt AMPure system (Agencourt Bioscience Corp., USA) before sequencing in an ABI 3730xl DNA Analyzer (Applied Biosystems, USA). Resulting sequences were compared with GenBank template sequences to identify the correct primate species, described in full in Frias et al. (Reference Frias, Stark, Lynn, Nathan, Goossens, Okamoto and MacIntosh2018).
Strongylid presence was assessed in each sample using HTS, where the entire strongylid community was determined by amplification of the second internal transcribed spacer (ITS-2) of nuclear DNA through PCR (polymerase chain reaction) using the forward primer Strongyl_ITS-2_F (ACGTCTGGTTCAGGGTTG) and the reverse Strongyl_ITS-2_R (ATGCTTAAGTTCAGCGGGTA) (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018). To generate HTS sequencing libraries a 2-step PCR approach that employs Nextera primer design was used (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018), with each sample tagged with a unique primer barcode, following Illumina libraries guide. Through the Illumina MiSeq platform the final library was sequenced using MiSeq Reagent Kit version 2 (2 × 250-bp pair-end reads, 500 cycles).
Bioinformatics and data analysis
Gene-specific primers were trimmed using SKEWER (Jiang et al., Reference Jiang, Lei, Ding and Zhu2014). Using DADA2, paired end reads were assembled (Callahan et al., Reference Callahan, McMurdie, Rosen, Han, Johnson and Holmes2016). The dataset was then denoised, low-quality reads (expected error rate >1) eliminated and inflation of strongylid diversity by PCR/sequencing artefacts (chimeras) avoided by corrections in DADA2 (Callahan et al., Reference Callahan, McMurdie, Rosen, Han, Johnson and Holmes2016). To taxonomically assign amplicon sequence variants (ASVs) the naïve Bayesian RDP classifier (Wang et al., Reference Wang, Garrity, Tiedje and Cole2007) was employed in the DADA2 pipeline. With strongylid ITS-2 sequences, downloaded from the NCBI nr/nt database (200 top BLASTN hits for each ASVs), a training database of reference sequences for assignment was constructed. The dataset was then filtered to remove all unclassified ASVs, those not assigned to Strongylida. Phylogenetic maximum likelihood analysis was performed using the web version of IQ TREE (Trifinopoulos et al., Reference Trifinopoulos, Nguyen, von Haeseler and Minh2016), with integrated ModelFinder selecting the most suitable model based on Bayesian information criterion (BIC) (Kalyaanamoorthy et al., Reference Kalyaanamoorthy, Minh, Wong, Von Haeseler and Jermiin2017), after removal of outlying ASV sequences with both low abundance and prevalence and addition of Oesophagostomum spp. sequences available in the GenBank. The topology of phylogenetic trees was tested using 1000 replicates of ultrafast bootstrap (Hoang et al., Reference Hoang, Chernomor, Von Haeseler, Minh and Vinh2018) and Shimodaira–Hasegawa approximate likelihood ratio test (SH-aLRT) (Anisimova et al., Reference Anisimova, Gil, Dufayard, Dessimoz and Gascuel2011). Through the model outputs, attention was also paid to pairwise distances between taxa, which were calculated from the alignments guided by Clustal Omega integrated into Geneious Prime 2023 2.1 (http://www.geneious.com). The trees were visualized and annotated in iTOL v5 (Letunic and Bork, Reference Letunic and Bork2021).
Differences in alpha diversity of strongylid communities were investigated by using the number of ASVs per sample as a proxy measure. Using the lme4 R package (de Boeck et al., Reference de Boeck, Bakker, Zwitser, Nivard, Hofman, Tuerlinckx and Partchev2011; Bates et al., Reference Bates, Mächler, Bolker and Walker2015), a GLMM (generalized linear mixed model) with negative binomial distribution was applied, accounting for aggregated parasite distributions, to assess the effect of host species on alpha diversity, including study site as a random factor. To assess overdispersion and ensure correct model selection, residual diagnostics were implemented and visually inspected through the DHARMa R package (Hartig, Reference Hartig2020), with no significant deviation found. Then the drop1() function (again in lme4 R package) was implemented to determine the importance of host species as a model effector, with strong significance identified comparative to a null hypothesis. Post hoc comparisons, using Tukey Honest Significant Differences, were employed to test the effect of host species factorially. Using the vegan R package (Oksanen et al., Reference Oksanen, Blanchet, Friendly, Kindt, Legendre, McGlinn, Minchin, O'Hara, Simpson, Solymos, Stevens, Szoecs and Wagner2019), between species variation in beta diversity, at the individual ASV level, was investigated through ANOSIM-based community compositional dissimilarities and tests of multivariate homogeneity, employing Bray–Curtis dissimilarities to account for ASV relative abundances. Clustering was visualized through weighted PCoA (principal coordinates analysis), again using the vegan R package (Oksanen et al., Reference Oksanen, Blanchet, Friendly, Kindt, Legendre, McGlinn, Minchin, O'Hara, Simpson, Solymos, Stevens, Szoecs and Wagner2019). To investigate within species variation of beta diversity, the same methods were applied to a data subset including only Japanese macaques, employing host locality as the effector variable.
Results
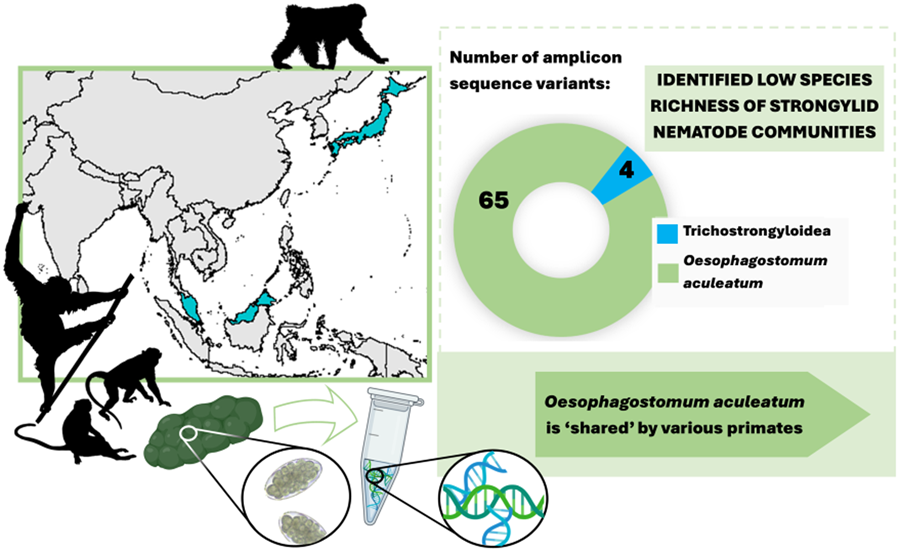
Sequencing data consisted of 4 554 168 ITS-2 reads, with an average of 50 602 (s.d. 62 306) reads per sample. In total, 69 individual ASVs were identified, 65 of which were assigned to a single strongylid species: Oesophagostomum aculeatum. The remaining 4 ASVs were unidentified as strongylid taxa, assigned to superfamily trichostrongyloidea and could not be assigned at the genus level. The pairwise sequence distance (PSD) among all Oesophagostomum ASVs derived from the samples reached up to 10%, although such differences were restricted to 6 ASVs (8, 50, 51, 52, 57 and 67), with the PSD among all other ASVs not exceeding 5.4%. In the phylogenetic tree, all ASVs clustered within one strongly supported clade corresponding to O. aculeatum, with further internal structure (Fig. 1). One subclade comprised ASVs found in Bornean orangutans at both localities in Sabah, with 2 of these ASVs also detected in Japanese macaques in Jigokudani. Moreover, GenBank sequences originating from orangutans, in both Borneo (P. pygmaeus) and Sumatra (P. abelii), and Bornean white-bearded gibbons (Hylobates albibarbis) clustered within this subclade. The PSD within this subclade was below 2.5%.

Figure 1. Maximum likelihood cladogram of Oesophagostomum ITS-2 region (245 bp), computed in IQ tree3 by model K2P + G4, using O. dentatum as an outgroup. The tree topology was tested by 1000 replicates of ultrafast bootstrap4 and SH-like aLRT5. Green circles mark nodes with support higher than 75%, with circle size depicting value. Sequences from GenBank are labelled by accession number.
Another small subclade comprised 5 ASVs (7, 9, 20, 23 and 30) detected in Japanese macaques across the 3 localities and one ASV (1) which occurred at all 3 Japanese localities as well as in crab-eating macaques at LKWS and orangutans at DVCA. This subclade also included 3 additional GenBank sequences from Japanese macaques. Other ASVs occurred across localities and host species, some occurred in multiple hosts at multiple localities, e.g. ASV 2 or ASV 22, some occurred in multiple hosts at a single locality, e.g. ASVs 25, 10 or 14, and some occurred in a single host at a single locality, e.g. ASVs 38, 27 or 69. Interestingly, these single-host-ASVs were always recorded in crab-eating macaques or Japanese macaques. It is worth mentioning that 2 sequences labelled as O. bifurcum in GenBank and originating from free-ranging bonobos (Pan paniscus) (Medkour et al., Reference Medkour, Castaneda, Amona, Fenollar, André, Belais, Mungongo, Muyembé-Tamfum, Levasseur, Raoult, Davoust and Mediannikov2021) clustered in the O. aculeatum clade and differed by only 1.2–6.6% (below 3.7% for most haplotypes) from any O. aculeatum sequence.
Of the 4 ASVs assigned only to trichostrongyloidea, all were restricted to a single host species at a single locality, occurring only in crab-eating macaques at LKWS, with none showing a close identity match with any GenBank sequences. All 4 ASVs clustered together and formed a small subclade (SUP FIG 1) closely related to a clade comprising Hyostrongylus spp. and to a clade including Ostertagia nianqingtanggulansis (AJ577459, from unspecified host from Tibet), Ostertagia sp. (AB367797, Japanese Serow, Japan), Mazamastrongylus dagestanica (JQ925868, OM445254; moose, Russia) and Spiculopteragia spp. (European cervids). Although M. dagestanica is currently assigned to the family Trichostrongylidae, all other taxa are from the family Haemonchidae, suggesting the nature of the trichostrongylids detected in macaques.
Upon visual inspection, alpha diversity (at the level of individual ASVs) of strongylid communities appeared to show minimal variation between host species (Fig. 2). GLMMs found statistically significant differences in alpha diversity with post hoc analysis revealing differences between only Japanese macaques and other host species, with no alpha diversity differences found among sympatric host species at LKWS (P > 0.08 for all pairwise comparisons). Japanese macaques showed lower alpha diversity of strongylid communities compared to crab-eating macaques, silvery lutungs and orangutans (P < 0.001 for all pairwise comparisons), yet no differences were found between the 3 localities in Japan. However, ASV composition of strongylid communities showed significant interspecific variation, both between host species (ANOSIM: R = 0.597, P = 0.001) and amongst Japanese macaques in different localities (ANOSIM: R = 0.425, P = 0.001), supported by visual inspection of ordination plots (Fig. 3).

Figure 2. Amplicon sequence variant (ASV) diversity of strongylid nematodes detected in faecal samples of South-East and East Asian primates from 5 localities, indicated across the upper x-axis.

Figure 3. Principal coordinate analysis (PCoA) plots of the beta diversity (assessed through Bray–Curtis dissimilarities) among strongylid nematode communities of (a) 6 South-East and East Asian Primate species from 5 localities and (b) M. fuscata from 3 localities in Japan. Convergent dots indicate similarities in community composition. While Koshima and Yakushima are represented by multiple samples, close convergence of points shows no separation within ordination space at this scale.
Discussion
Faecal samples from 6 NHP species inhabiting regions of South-East and East Asia were examined to assess the diversity of their strongylid nematode communities. A community profiling approach was applied, based on a previously optimized and tested HTS amplicon sequencing methodology. Focusing on strongylid communities from multiple NHP species allowed the diversity of these communities to be investigated with reference to sympatric species, different localities and host phylogenies. The reported diversity of strongylid nematodes highlights the potential consistency of these communities in NHP hosts within South-East and East Asia. The limitations of HTS methods should be noted, most poignantly the restriction in taxonomic resolution due to short sequence lengths and potential for sequencing errors from PCR biases (Kircher and Kelso, Reference Kircher and Kelso2010; Ambardar et al., Reference Ambardar, Gupta, Trakroo, Lal and Vakhlu2016). However, previous work (Pafčo et al., Reference Pafčo, Čížková, Kreisinger, Hasegawa, Vallo, Shutt, Todd, Petrželková and Modrý2018) has shown successful approaches for diminishing the impact of these stochastic effects.
There was a clear dominance of O. aculeatum, with nearly all ASVs assigned to this taxon. Dominance of O. aculeatum was expected, mirroring previous reports of strongylid infections in Asian NHPs (Frias et al., Reference Frias, Stark, Salgado Lynn, Nathan, Goossens, Okamoto and MacIntosh2019). Still, the near complete absence of other strongylid taxa was surprising, as strongylid nematodes occur in complex communities comprised of multiple genera in other studied hosts. The majority of studied samples showed single species infections, contrasting with reports of strongylid infections in African NHPs (Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019; Mason et al., Reference Mason, Petrzelkova, Kreisinger, Bohm, Cervena, Fairet, Fuh, Gomez, Knauf, Maloueki, Modry, Shirley, Tagg, Wangue and Pafco2022; Ilík et al., Reference Ilík, Kreisinger, Modrý, Schwarz, Tagg, Mbohli, Nkombou, Petrželková and Pafčo2023). Previously implemented molecular methods, involving individual larval specimens, identified the presence of a single larva of Ternidens in one Bornean orangutan (Frias et al., Reference Frias, Stark, Salgado Lynn, Nathan, Goossens, Okamoto and MacIntosh2019), raising questions as to why this study did not identify the presence of Ternidens. The absence of Necator americanus among these samples should also be noted, as this strongylid has previously been reported in the local human population (Lim-Leroy and Chua, Reference Lim-Leroy and Chua2020) and is known to be frequently transmitted between humans and wild NHPs in Africa (Pafčo et al., Reference Pafčo, Kreisinger, Čížková, Pšenková-Profousová, Shutt-Phillips, Todd, Fuh, Petrželková and Modrý2019). Potentially, this study is restricted by the limited sample size of some host species (especially orangutans at LKWS), with further sampling required for a more complete picture of strongylid diversity among Asian NHPs.
ASVs of O. aculeatum found in this study were further separated into 2 clusters. The first cluster consisted mainly of ASVs from orangutans, with 2 additional ASVs identified from Japanese macaques. This cluster also includes 4 sequences from GenBank, recorded as infecting P. pygmaeus, P. abelii and H. albibarbis, suggesting a lineage specialized to orangutans, with historical spill-over to other hosts. This cluster assembled closely with the 2 sequences from bonobos registered as O. bifurcum, though based on clustering these sequences may in fact represent O. aculeatum (Medkour et al., Reference Medkour, Castaneda, Amona, Fenollar, André, Belais, Mungongo, Muyembé-Tamfum, Levasseur, Raoult, Davoust and Mediannikov2021). The second cluster contained more ASVs and is identified in a larger diversity of host species, indicating it may be a more generalist lineage. Differentiation of these clusters as lineages or species would require morphological examination of adult worms and analysis of more markers. Cryptic species of Oesophagostomum have previously been proposed for nodular worm infections in humans and NHPs in Uganda (Ghai et al., Reference Ghai, Chapman, Omeja, Davies and Goldberg2014), with high genetic variability of Oesophagostomum spp. observed in African primates (Sirima et al., Reference Sirima, Bizet, Hamou, Červená, Lemarcis, Esteban, Peeters, Mpoudi Ngole, Mombo, Liégeois, Petrželková, Boussinesq and Locatelli2021; Mason et al., Reference Mason, Petrzelkova, Kreisinger, Bohm, Cervena, Fairet, Fuh, Gomez, Knauf, Maloueki, Modry, Shirley, Tagg, Wangue and Pafco2022). However, although the ITS-2 marker, also used in this study, is ideal for distinguishing between known species for which genetic data exists in repositories, it should be noted that this marker shows high levels of intraspecific variability, which may limit its value in identifying potential cryptic species (Conole et al., Reference Conole, Chilton, Järvis and Gasser1999; Poissant et al., Reference Poissant, Gavriliuc, Bellaw, Redman, Avramenko, Robinson, Workentine, Shury, Jenkins, McLoughlin, Nielsen and Gilleard2021; Halvarsson and Tydén, Reference Halvarsson and Tydén2023). Several host-specific lineages were previously found in Oesophagostomum following cox1 analyses with 2 clear lineages showed within O. aculeatum, the first being found in a number of hosts, while the second being more host-specific (Frias et al., Reference Frias, Stark, Salgado Lynn, Nathan, Goossens, Okamoto and MacIntosh2019). This confirms a certain degree of host preference within the O. aculeatum lineages detected and shows that involving genetic markers of divergence at the mitochondrial DNA loci would be an important aid in deciphering potentially cryptic species (Blouin, Reference Blouin2002).
The 4 trichostrongyloidea ASVs identified in crab-eating macaques highlight the current convolution in genetic distinction of strongylids in wildlife hosts. There are records of unspecified Trichostrongylus sp. based on the presence of eggs in various Asian macaque species (Kumar et al., Reference Kumar, Sundararaj, Kumara, Pal, Santhosh and Vinoth2018; Kurniawati et al., Reference Kurniawati, Kurniawati, Suwanti, Lastuti, Kusdarto, Suprihati, Mufasirin and Pratiwi2020; Fernando et al., Reference Fernando, Udagama and Fernando2022). Although humans and some African cercopithecoid primates are occasionally reported with Trichostrongylus colubriformis infection (Phosuk et al., Reference Phosuk, Intapan, Sanpool, Janwan, Thanchomnang, Sawanyawisuth, Morakote and Maleewong2013; Sharifdini et al., Reference Sharifdini, Derakhshani, Alizadeh, Ghanbarzadeh, Mirjalali, Mobedi and Saraei2017; Obanda et al., Reference Obanda, Maingi, Muchemi, Ng'Ang’A, Angelone and Archie2019), and apparently there are Trichostrongylus spp. infecting African great apes (Mason et al., Reference Mason, Petrzelkova, Kreisinger, Bohm, Cervena, Fairet, Fuh, Gomez, Knauf, Maloueki, Modry, Shirley, Tagg, Wangue and Pafco2022), these sequences did not cluster with any Trichostrongylus spp. provided in the GenBank. Thus, as a more probable candidate Nochtia nochti is proposed, a trichostrongyloid nematode parasitizing Macaca spp. across Asia which unfortunately is not represented in available sequence databases. However, pathogenesis described in N. nochti (Yalcindag et al., Reference Yalcindag, Stuart, Hasegawa, Streit, Doležalová, Morrogh-Bernard, Cheyne, Nurcahyo and Foitová2021; Fonti et al., Reference Fonti, Parisi, Mancianti, Freer and Poli2023) holds similarities to other trichostrongyloids from the family Haemonchidae (Deplazes et al., Reference Deplazes, Eckert, Mathis, von Samson-Himmelstjerna and Zahner2016), within which these sequences clustered.
The number of strongylid ASVs per sample, a quasi-measure of alpha diversity, was generally consistent across host species. Of all host species, only Japanese macaques showed noticeable differences (significantly lower number of ASVs) from other species. This finding may be as expected, with Japanese macaques being the unique sampled species occurring in East Asia, compared to the other species in South-East Asia. Of particular note is the variation identified between the 2 macaque species, Japanese macaques and crab-eating macaques, highlighting how geographical factors and sympatric species can influence strongylid communities of closely related host taxa. Japanese macaques are the world's most northern-living primate (Cozzolino et al., Reference Cozzolino, Cordischi, Aureli and Scucchi1992; Tsuji et al., Reference Tsuji, Kazahari, Kitahara and Takatsuki2008), meaning it is one of the few NHP species that exists solely in single-primate communities (Ito et al., Reference Ito, Hayakawa, Suzuki–Hashido, Hamada, Kurihara, Hanya, Kaneko, Natsume, Aisu, Honda, Yachimori, Anezaki, Omi, Hayama, Tanaka, Wakamori, Imai and Kawamoto2021). As such, the ability for parasites to host switch, a method whereby a parasite establishes within a novel host species (De Vienne et al., Reference De Vienne, Giraud and Shykoff2007; Cooper et al., Reference Cooper, Griffin, Franz, Omotayo and Nunn2012), is limited due to the absence of closely related species already hosting the parasite. Fossil evidence suggests that Japanese macaques may have long existed within a single-primate community (Marmi et al., Reference Marmi, Bertranpetit, Terradas, Takenaka and Domingo-Roura2004; Kawamoto et al., Reference Kawamoto, Shotake, Nozawa, Kawamoto, Tomari, Kawai, Shirai, Morimitsu, Takagi, Akaza, Fujii, Hagihara, Aizawa, Akachi, Oi and Hayaishi2007), which may also be reflected among their intestinal parasite fauna (Gotoh, Reference Gotoh2000).
Additionally, this northern distribution means Japanese macaques inhabit Japanese islands where environmental conditions differ quite dramatically from those in Borneo. Japanese macaques living at more northern latitudes, where the climate is characterized by colder winters, were previously shown to carry fewer gastrointestinal parasites at lower prevalence (Gotoh, Reference Gotoh2000). The harsher seasonal environment in Japan may reduce the longevity of strongylids during developmental stages in the external environment (Knapp-Lawitzke et al., Reference Knapp-Lawitzke, Küchenmeister, Küchenmeister, von Samson-Himmelstjerna and Demeler2014). As such, ASV diversity, in Japan compared to Borneo, is limited by the robustness of strongylid taxa and their ability to withstand harsher environmental conditions.
Great variation between the samples according to the community composition of strongylids was observed both among species as well as within species (when concerning Japanese macaques). Some of this among-species variation is explained by the 2 identified lineages of O. aculeatum, with the divergent ordination of orangutans coinciding with the ‘orangutan’ lineage, almost exclusively identified within this host. Likewise, the divergent ordination of crab-eating and pig-tailed macaques from Japanese macaques attests to the importance of sharing a habitat with other sympatric primates, supported by the widespread second lineage of ASVs identified within these hosts. Japanese macaques occupying a separate ordination space are additionally at least in part due to the reduced diversity of strongylid communities in this host species. This divergent ordination of macaque species, suggesting divergent composition of strongylid communities, contradicts the host–parasite co-speciation hypothesis (Brooks, Reference Brooks1979). Interestingly, only a single ASV was commonly identified among all 3 macaque species, with Japanese macaques sharing just 3 additional ASVs with either crab-eating or pig-tailed macaques, highlighting the role of environmental factors influencing parasite communities. Divergent ordination among Japanese macaques from different localities further emphasizes this, with hosts of the same species showing variation within strongylid community composition and only a single shared ASV identified across all 3 localities. Geographical isolation within Japanese macaques has led to the evolution of a subspecies in one locality: M. f. yakui in Yakushima (Hayaishi and Kawamoto, Reference Hayaishi and Kawamoto2006). Host evolution is not mirrored by their strongylid communities in this case, however, with Yakushima strongylid communities showing convergence, in terms of composition, with Koshima, while Jigokudani strongylid communities show far greater divergence, again opposing the host–parasite co-speciation hypothesis. Further exploration of this hypothesis among NHPs, and environmentally driven deviation, could benefit from using the Macaca genus as a model, with 22 currently recognized species distributed into several lineages (Thierry, Reference Thierry2007) and occupying the widest geographical range of all NHP, spanning both tropical and temperate regions (Fooden, Reference Fooden1982).
Concluding, the strongylid nematode communities of South-East and East Asian NHP were dominated by O. aculeatum, with single-species occurrence identified in most samples, which is in sharp contrast to the rich strongylid communities harboured by African great apes and other African NHPs. The genetic distances between sequences suggests the potential for 2 cryptic lineages within O. aculeatum. One lineage appears more specialized in terms of host diversity, with the other being more generalist and diverse. However, ITS-2 does not provide a suitable marker for evaluation of intraspecific diversity, therefore further sequencing with the inclusion of more genetic markers is required to better decipher the genetic make-up of the O. aculeatum complex. These findings highlight how geographic variables may influence parasite communities within NHPs, disuniting host–parasite co-speciation. While the reported strongylid communities seem low in species richness, this report is only an initial insight and additional sampling is required to truly capture the genetic diversity of strongylid communities in Asian NHPs. Establishing baseline data on NHP strongylid infections can assist with ongoing monitoring of health threats to wild NHPs with HTS providing complex insights into strongylid community composition and ability to detect even less prevalent, but more pathogenic taxa.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182024000386.
Data availability statement
Strongylid ITS-2 sequencing data is archived in the European Nucleotide Archive, under the accession number of the whole project PRJEB73875 (available at: https://www.ebi.ac.uk/ena/browser/view/PRJEB73875). Accession numbers for each sample are available alongside related metadata in Supplementary Table 1, which includes geographical location and sampling period.
Acknowledgements
The authors extend gratitude to the Sabah Wildlife Department and Sabah Biodiversity Centre (SaBC) for allowing us to conduct this research through multiple access licences (JKM/MBS.1000-2/3 (101) to RM; JKM/MBS.1000-2/2 (280), JKM/MBS.1000-2/2 JLD.3 (106), JKM/MBS.1000-2/2 JLD.5 (82), JKM/MBS.1000-2/2 JLD.6 (121) to LF; JKM/MBS.1000-2/2 JLD.3 (35), JKM/MBS.1000-2/2 JLD.5 (68), JKM/MBS.1000-2/2 JLD.6 (108) to AJJM) and sample export licences (JKM/MBS.1000-2/3 JLD.2 (62) to AJJM; JKM/MBS.1000-2/3 JLD.3 (108) to SP), as well as to Yayasan Sabah for permission to work at DVCA. We also thank Noko Kuze, Tomoko Kanamori, Henry Bernard and Peter Malim for supporting research at DVCA. We thank the Cooperative Research Programme of the Wildlife Research Center and Kushima City for permissions to conduct research at Koshima, while also thanking the Cooperative Research Programme of the Wildlife Research Center, Yakushima Forest Office and Kagoshima Prefecture for permission to conduct research on Yakushima. Additionally, we thank Jigokudani Monkey Park for permission to conduct research there.
Author contributions
AJJM, BP and KJP conceived and designed the study while LF, BG, KK, AL, RM, SP, SS, ZX and AJJM collected and/or facilitated sample collection. BM, KK, AL, KMaj and ZX undertook initial laboratory work, under supervision of MO, BP and AJJM. HH, KK, AL, TM, KMaj, KMat, ZX and BP were responsible for sequencing work, with the resulting data then analysed by BM, BC and BP. BM wrote the article initially, with supervision from BP, AJJM and KJP, before all co-authors reviewed and improved the article.
Financial support
We are extremely grateful to the Operational Programme Research, Development and Education – Project Internal Grant Agency of Masaryk University (No. CZ.02.2.69 / 0.0 / 0.0/ 19_073/ 0016943) for supporting this work and in particular funding BM's involvement. We are also grateful for the support from Japan Society for the Promotion of Science (JSPS)-Czech Academy of Science (JSPS-CAS) bilateral cooperative programme, Japan/Czech Republic (project Nos. JPJSBP120192506 and JSPS-19-17) and from the Czech Science Foundation (22-16475S). We extend further gratitude to JSPS for supporting our research in Borneo, through grants awarded to AJJM (#24770232, #20H03333, #16H06181), MO (#15H04283) and LF (#446). The study was further supported through Kyoto University's Step-Up Program awarded to AJJM. Finally, we thank the Cooperative Research Programme of the Kyoto University Primate Research Institute, for supporting SP's involvement.
Competing interests
The authors declare there are no conflicts of interest.
Ethical standards
Authorization to conduct research and collect biological samples in Sabah was granted by the Sabah Biodiversity Centre (SaBC) and Yayasan Sabah, with the support of the Sabah Wildlife Department. Our field protocols strictly followed the guidelines established by these agencies, as well as the regulations set by the Field Research Committee at the former Kyoto University Primate Research Institute, Japan.






 Open access
Open access