



Introduction
Schistosomiasis is a neglected tropical disease caused by platyhelminths of the genus Schistosoma. The disease has global impact on human and animal health. According to the WHO, approximately 600 million people live in endemic areas, of which >200 million require treatment with a mortality rate of ~24 000 people in 2016 (WHO Global, 2016). Schistosomes belong to the few members of platyhelminths that have evolved separate sexes (Beltran and Boissier, Reference Beltran and Boissier2008), and they exhibit a unique reproductive biology because the female's sexual maturation depends on a constant pairing-contact with the male (Kunz, Reference Kunz2001; LoVerde, Reference LoVerde2002). Currently medical treatment is based upon a single drug, praziquantel. Although it is safe, affordable and effective, its widespread use for mass treatment has increased the likelihood of resistant parasites (Bergquist et al., Reference Bergquist, Utzinger and Keiser2017). This requires the development of alternative antischistosomal compounds.
G protein-coupled receptors (GPCRs) are the largest known superfamily of membrane receptors with a seven-transmembrane domain, and they typically interact with ligands in the extracellular domain and with heterotrimeric GTP binding proteins (G proteins) at intracellular domains. GPCRs are phylogenetically classified into five families, Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin (Schiöth and Fredriksson, Reference Schiöth and Fredriksson2005). Neuropeptides typically activate receptors from the Rhodopsin family (class A receptors). Upon ligand binding, a GPCR undergoes conformational changes and releases membrane-associated G-proteins (subunits α, β and γ). In this state, the activated GPCR acts as a guanine nucleotide exchange factor by inducing GTP (guanosine triphosphate) exchange in the α subunit and its consequent dissociation from the βγ dimer; both dissociated α and βγ subunits can elicit downstream effects (Frooninckx et al., Reference Frooninckx, van Rompay, Temmerman, van Sinay, Beets, Janssen, Husson and Schoofs2012).
The neuropeptidergic signalling component of invertebrate nervous systems is highly developed and complex. The system involves many different neuropeptide components, the neuropeptide processing enzymes, the endogenous neuropeptide receptors and further molecules involved in signalling cascades. Neuropeptide maturation occurs in a modulated fashion in helminths compared to vertebrates (Mair et al., Reference Mair, Niciu, Stewart, Brennan, Omar, Halton, Mains, Eipper, Maule and Day2004; Atkinson et al., Reference Atkinson, McVeigh, Kimber, Marks, Eipper, Mains, Day and Maule2010); a single gene can encode multiple copies of the same or different neuropeptides, and the precursor molecule undergoes extensive posttranslational processing in the trans-Golgi network prior to the production of individual neuropeptides. The processing in trematodes involves three major steps, (1) digestion of a precursor protein by a prohormone convertase (PC) at specific dibasic amino acid motifs (often two consecutive residues of lysine and arginine), (2) digestion of the carboxyterminal amino acid (aa) of the peptide-processing intermediates by a carboxypeptidase E (CPE), and (3) the amidation of the glycine C-termini in two sequential steps by peptidylglycine-α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL) (McVeigh et al., Reference McVeigh, Mair, Novozhilova, Day, Zamanian, Marks, Kimber, Day and Maule2012).
In Schistosoma mansoni, 47 potential neuropeptides emerging from 32 neuropeptide precursor (npp) genes have been identified by in silico analysis (McVeigh et al., Reference McVeigh, Atkinson, Marks, Mousley, Dalzell, Sluder, Hammerland and Maule2009; Koziol et al., Reference Koziol, Koziol, Preza, Costábile, Brehm and Castillo2016). Furthermore, RNAseq analysis provided a comprehensive dataset of genes that are differentially transcribed in paired and unpaired schistosome males and females (Lu et al., Reference Lu, Sessler, Holroyd, Hahnel, Quack, Berriman and Grevelding2016). Among these were 27 Sm_npps, and all major subfamilies of GPCRs including a platyhelminth-specific rhodopsin subfamily (Zamanian et al., Reference Zamanian, Kimber, McVeigh, Carlson, Maule and Day2011; Campos et al., Reference Campos, Young, Korhonen, Hall, Mangiola, Lonie and Gasser2014; Hahnel et al., Reference Hahnel, Wheeler, Lu, Wangwiwatsin, McVeigh, Maule, Berriman, Day, Ribeiro and Grevelding2018). Interestingly, transcript levels of almost all Sm_npps and many GPCRs are highest in males (paired or unpaired) and also in unpaired females. This suggested lower importance of neuronal processes in the female after pairing (Lu et al., Reference Lu, Spänig, Weth and Grevelding2019).
Our knowledge of the biological meaning of neuronal signalling via GPCRs is still fragmentary in schistosomes. Unravelling these processes would help to understand the biology of the parasite and at least part of the interplay between males and females. Towards this end, the identification of endogenous ligands for trematode peptidergic GPCRs (deorphanization) is one step needed to enable the functional characterization of GPCRs.
The primary aim of this work was to find a way to deorphanize S. mansoni GPCRs by employing the Membrane-Anchored Ligand And Receptor yeast two-hybrid system (MALAR-Y2H) system. As a state of the art method, this system broadens the spectrum of technical possibilities to identify protein–protein interactions (PPIs) of transmembrane receptors and extracellular ligands (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). To achieve this goal we generated a library of 47 neuropeptides fused via a flexible linker to the transmembrane domain (TMP) of yeast WBP1 (te Heesen et al., Reference te Heesen, Rauhut, Aebersold, Abelson, Aebi and Clark1991). The intracellular part of these fusion constructs consisted of the C-terminal part of ubiquitin (Cub) and the transcription factor GAL4. The N-terminal part of ubiquitin (NubG) is joined to another construct harbouring the potential interaction partner, a GPCR. Interaction of ligands and receptors initiate a cascade that ultimately liberate the transcription factor GAL4 leading to reporter gene activation. Here, we present the full-length expression of seven S. mansoni GPCRs in a heterologous yeast system. One of these coding sequences (CDS) had to be codon-optimized to meet the requirements of the yeast 3′ processing machinery during transcription (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015). Furthermore, we detected fusions of (1) ligands with TMP and (2) receptors, linked on the C-terminus with GFP, and localized these to the plasma membrane of yeast cells. The results of the MALAR-Y2H assay suggested neuropeptide-receptor interaction of two GPCRs, which allowed us to identify potential ligands.
Materials and methods
Plasmid constructs
Based on previous work, we generated bait plasmid pGAD SP-WBP1_cloning_linker_TMP_Cub_ GAL4 by cutting pGAD WBP1-Cub-GAL4 (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016) with KpnI and MluI and assembled it with a gBlock® (IDT, Iowa, USA) to destroy Acc65I restriction site. The intermediate clone was digested with NcoI, and a second gBlock was introduced with the following modifications: (1) WBP1 aa 24-430 was deleted, (2) a flexible linker was introduced 5′ to TMP, and (3) new Acc65I and SmaI restriction sites were generated to insert neuropeptides in frame with the WBP1 signal-peptide aa 1-23.
The neuropeptide library was generated by cutting pGAD SP-WBP1_cloning_linker_TMP_Cub_ GAL4 with Acc65I and SmaI, and by ligating annealed primers (Supplementary Table 1) with corresponding 3′ and 5′ overhangs. pGAD SP-WBP1_cloning_linker_TMP_eGFP for subcellular localization studies was constructed by cutting pGAD SP-WBP1_cloning_ linker_TMP_Cub_GAL4 with SacI and HindII to remove Cub and GAL4. The coding sequence for eGFP was introduced using Gibson Assembly (NEB, MA, USA) and accordingly, neuropeptides were cloned by primer annealing.
Prey plasmids were created by cutting pGBKT7 OST1-NubG with NcoI and NotI and assembled it with a gBlock utilizing Gibson Assembly. The resulting vector pGBKT7_SP_OST1_ cloning_NubG had the following modifications: (1) OST1 aa 28-481 were deleted retaining the SP of OST1, (2) replacement of existing NcoI by the same restriction site after SP of OST1, (3) introducing a SmaI restriction site in frame 5′ to NubG. The CDS of GPCR-coding genes were amplified, using Q5® polymerase, from cDNA obtained from total RNA preparation of S. mansoni. Primers were designed to assemble products into pGBKT7_SP_OST1_cloning_NubG cut with NcoI and SmaI. For subcellular localization studies, NubG was replaced by eGFP.
Cell lines
Escherichia coli strain NEB® 10-beta was used to clone and amplify plasmid DNA. Yeast strains used for mating-assay were AH109 (MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATAHIS3, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1 UASMEL1TATA-lacZ, MEL1) and Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met–, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ) from Clontech (Matchmaker® Y2H-system).
RNA isolation, cDNA synthesis and polymerase chain reaction (PCR)
Rna was isolated from adult schistosomes or yeast clones with the Monarch® Total RNA Miniprep Kit (NEB) and transcribed into cDNA using ProtoScript® II reverse transcriptase (NEB). Both steps were carried out following the manufacturer instructions. PCR analysis for cloning full-length GPCR coding sequences and expression in yeast was performed with primer pairs as indicated in Supplementary Table 2. In brief, amplification was done with Q5 High-Fidelity DNA polymerase in a total volume of 25 μL for 32 cycles (95°C for 8 s, 58°C for 20 s and 72°C for 100 s). Amplified PCR products were resolved by agarose gel electrophoresis.
Yeast transformation and mating
Transformation of Saccharomyces cerevisiae yeast strains was performed according to Tripp et al. (Reference Tripp, Lilley, Wood and Lewis2013) utilizing an optimized LiAc-method. Prey plasmids were transformed into Y187 strain and bait plasmids were transformed into AH109 strain. Transformed yeasts were selected by plating on synthetic dropout medium (SD) lacking tryptophane (SD/Trp−) or leucine (SD/Leu−), respectively. Mating was achieved through resuspending 10 μL of each AH109 and Y187 clone in 500 μL YPDA medium and cultivated for 16 h at 30°C. Hybrids carrying both plasmids were selected on SD/Trp− Leu− plates. Growth assay to monitor PPI was carried out by plating a dilution series (OD600 = 1, 0.1 and 0.01) of two mated yeast clones (n = 2) on SD/Trp− Leu− His− Ade−. Interaction was documented after 72 h at 30°C by scanning the plates.
β-Galactosidase assay
Colonies of mated yeasts were cultivated in SD/Trp− Leu−; medium until OD600 of 0.4–0.8 was reached. One millilitre culture medium was centrifuged, and pelleted cells were resuspended in 400 μL Z-buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, pH 7.0). Cells were lysed by three consecutive freeze/thaw cycles in liquid nitrogen. The lysate was mixed with 200 μL Z buffer containing 0.4% o-nitrophenyl-β-d-galactopyranoside (ONPG) and incubated for 30 min at 30°C. After centrifugation, the absorbance of the supernatant at 405 nm was measured and Miller Units were calculated according to the equation: Miller Units = 1000 × OD405/t(min) × OD600.
Confocal laser scanning microscopy
For localization analysis of receptors and neuropeptides by confocal laser scanning microscopy (CLSM), yeast cells were transfected with GFP fusion constructs and stained with the membrane specific dye Dil (Merck KGaA, Germany). In brief, cells were fixed for 10 min on ice with 4% paraformaldehyde dissolved in PBS. After two washing steps, cells were incubated with 10 μ m Dil and washed four times with PBS. Finally, yeasts were dried and mounted with Roti®-Mount FluorCare (Carl Roth, Germany). Stained yeast cells were captured on an inverse CLSM (Leica TSC SP5; Leica, Germany). GFP was excited with an argon-ion laser at 488 nm and Dil at 561 nm.
Maintenance of the schistosome life cycle
A Liberian strain (Bayer AG, Monheim) of S. mansoni was used to infect freshwater snails of the genus Biomphalaria glabrata as an intermediate host and Syrian hamsters (Mesocricetus auratus) as final host (Grevelding, Reference Grevelding1995). Eight-week-old hamsters were infected by the ‘paddling method’ (Dettman et al., Reference Dettman, Higgins-Opitz and Saikoolal1989), and sacrificed at 46 days post infection to collect adult worm couples by perfusion. Worms were cultured in M199 medium [Sigma-Aldrich, Germany; supplemented with 10% Newborn Calf Serum, 1% HEPES (1 M) and 1% ABAM-solution (10 000 units penicillin, 10 mg streptomycin and 25 mg amphotericin B per mL)] at 37°C and 5% CO2.
Ethical standard
Animal experiments were performed in accordance with the European Convention for the Protection of Vertebrate Animals used for experimental and other scientific purposes (ETS No 123; revised Appendix A) and were approved by the Regional Council (Regierungspraesidium) Giessen (V54-19 c 20/15 c GI 18/10).
Results
Modulation of the MALAR-Y2h system
The MALAR-Y2H system is a further development of the split ubiquitin method to detect protein–protein interactions between two membrane proteins (Stagljar et al., Reference Stagljar, Korostensky and Johnsson1998; Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). Y2H analysis contributed significantly to identify interacting proteins in the past with the limitation to cytosolic and membrane-bound proteins. The MALAR-Y2H method expands these applications through a chimeric transmembrane protein allowing the analysis of membrane-bound proteins and their putative interactions. In this split ubiquitin-based system, one potential interaction partner is expressed as a chimeric transmembrane protein, of which one part is an extracellular ligand fused via a transmembrane domain to an intracellular part. The latter consists of Cub and the transcription factor GAL4. The other potential interaction partner is the receptor of interest that is fused to NubG. The principle of the MALAR-Y2H system to identify PPIs is the reconstitution of the two ubiquitin halves, which is triggered by ligand–receptor interaction. Only re-formed ubiquitin can be recognized and cleaved by an endogenously expressed ubiquitin-specific protease (UBP). As a consequence GAL4 is released and transported to the nucleus to induce reporter gene expression (Fig. 1).
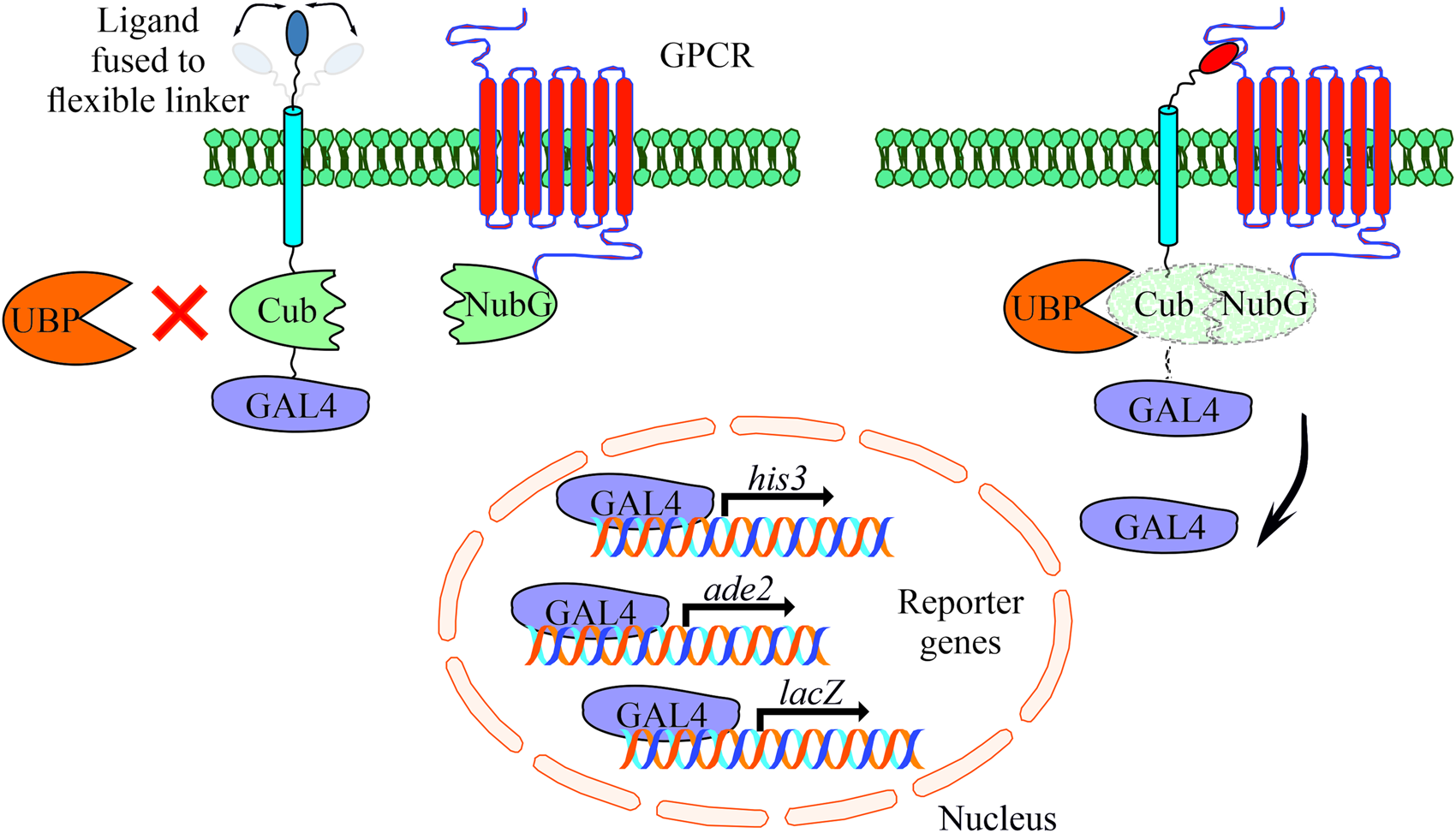
Fig. 1. Schematic representation of the yeast-based MALAR system for detecting interactions of extracellular ligands with their potential membrane receptors. The ligands are expressed as fusion proteins and consist of a flexible linker directed to the extracellular space, a transmembrane domain, the C-terminal part of ubiquitin (Cub), and the GAL4 transcription factor. Receptors are fused to the N-terminal part of ubiquitin (NubG) harbouring a single point mutation (Ile13Gly) to prevent spontaneous reconstitution. These vectors complement leucine and tryptophan auxotrophs, respectively. An interaction of ligand and receptor forces the split ubiquitin halves into close proximity. A ubiquitin-specific protease (UBP) recognizes the reconstituted ubiquitin, which is subsequently degraded leading to the release of GAL4. The latter enters the nucleus activating the reporter genes his3, ade2 and lacZ to allow cell growth on selective agar plates (SD/Leu− Trp− His− Ade−).
To further improve and adapt the system to our requirements, we introduced a flexible linker between the transmembrane domain and the neuropeptide (Argos, Reference Argos1990). Most of the mature neuropeptides of schistosomes are between four and twenty amino acids in size (Koziol et al., Reference Koziol, Koziol, Preza, Costábile, Brehm and Castillo2016), thus they represent relatively small molecules compared to classic chemokines like the CXC-motif chemokine ligand 12 (CXCL12) that was used in the study of Li et al. (Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). Flexible linkers consist of relatively small amino acids such as glycine, serine and threonine and often occur in natural proteins. Besides their flexibility, they are polar to form hydrogen bonds with water helping to maintain the stability of the linker structure in aqueous solvents. This may prevent steric hindrance due to a rigid structure of the chimeric transmembrane protein, and we assumed that this may facilitate ligand–receptor interaction.
Full-length expression of S. mansoni GPCRs in S. cerevisiae
As shown in a previous study, AT-rich DNA sequences of S. mansoni genes can be misinterpreted by the yeast 3′ processing machinery during transcription as cleavage and poly-adenylation signals, which leads to truncated transcripts (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015). To overcome this limitation, a computer program was developed for the prediction of mRNA 3′ processing sites by providing a discrete state-space (DSM) value (Graber et al., Reference Graber, McAllister and Smith2002). A higher DSM value indicates a higher probability of a 3′ processing site at a specific DNA sequence. For S. mansoni α integrins 2, 3 and 4 it was shown that DSM values >14 exhibited a 3′ processing signal leading to truncated proteins (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015). We addressed this by examining full-length transcription after cloning GPCR coding sequences into a yeast expression vector, which were transformed into strain Y187. Total RNA was isolated and reverse transcribed into cDNA (+RT) followed by a PCR using specific primers for amplifying full-length transcripts of respective GPCRs.
Of seven GPCR-coding genes, only for one (Smp_244240) no amplification product was obtained using gene-specific primers (data not shown). This correlated with maximum DSM values up to 20.58 for this gene. PCR analyses with three different primer pairs amplifying the 5′-proximal region of Smp_244240 (Supplementary Fig. 1A) resulted in an amplification product using reverse primer 1 (rev1) only in one out of several yeast clones tested. No products were obtained using other primers rev2 or rev3. It appeared that the mRNA was prematurely truncated, probably between base pairs 190 and 301. This assumption coincided with the highest DSM peaks for Smp_244240, which occurred between base pairs 200 and 350 (Supplementary Fig. 1B). Next, we applied the Java codon adaption tool (JCAT) as described before (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015) to optimize the coding sequence without affecting the amino acid sequence. The resulting product was sequenced to confirm identity before subsequent cloning into the yeast expression vector.
The optimized sequence (Smp_244240 opt) and the other six GPCRs (DSM scores <13) tested showed full-length transcripts in yeast (Fig. 2). No signal was detected in the absence of reverse transcriptase (−RT), which excluded DNA contamination of the RNA samples. Reverse transcribed RNA isolated from S. mansoni showed the same size of amplicons as demonstrated for the yeast clones. These results suggested that full-length transcripts of these GPCRs were present in yeast.

Fig. 2. Full-length expression of S. mansoni GPCRs in S. cerevisiae. Wild type or optimized CDSs of GPCRs were transformed into yeast strain AH109. RNA was isolated and transcribed into cDNA. Specific primer pairs were used to amplify corresponding GPCRs (numbers 1-7) by RT-PCR, and resulting amplicons were size-separated on a 1% agarose gel (M = marker, bp = base pair). The absence of reverse transcriptase (-RT) during cDNA synthesis or cDNA synthesis without template (H2O) served as negative controls. RNA isolated from S. mansoni served as a positive control for full-length transcripts. In each case, amplicons of the expected sizes were obtained.
Detecting plasma membrane localization of heterologous GPCRs in S. cerevisiae
To reveal the cellular localization of recombinant S. mansoni GPCRs guided by the signal peptide of OST1 (yeast OST1, residues 1–27) in yeast, we tagged selected GPCRs at their C-terminus with eGFP. Microscopic expression analysis revealed that GPCR9 (Smp_244240) and GPCR14 (Smp_203500) were predominantly located in the membrane as visualized by an overlay with the membrane-specific dye Dil (Honig and Hume, Reference Honig and Hume1986) (Fig. 3). In contrast, the fluorescent signals for GPCR13-GFP (Smp_128170) were mainly detected in the cytoplasm (Supplementary Fig. 2B), as expected for the expression of GFP without fusion partner. Furthermore, besides their cytoplasmic occurrence, we confirmed that fusion proteins of a representative neuropeptide (NPP5a), fused to the transmembrane domain WBP1 (yeast WBP1, residues 341–430) and guided by the signal peptide of WBP1 (residues 1–23), was also targeted to the outer membrane (Supplementary Fig. 2A).
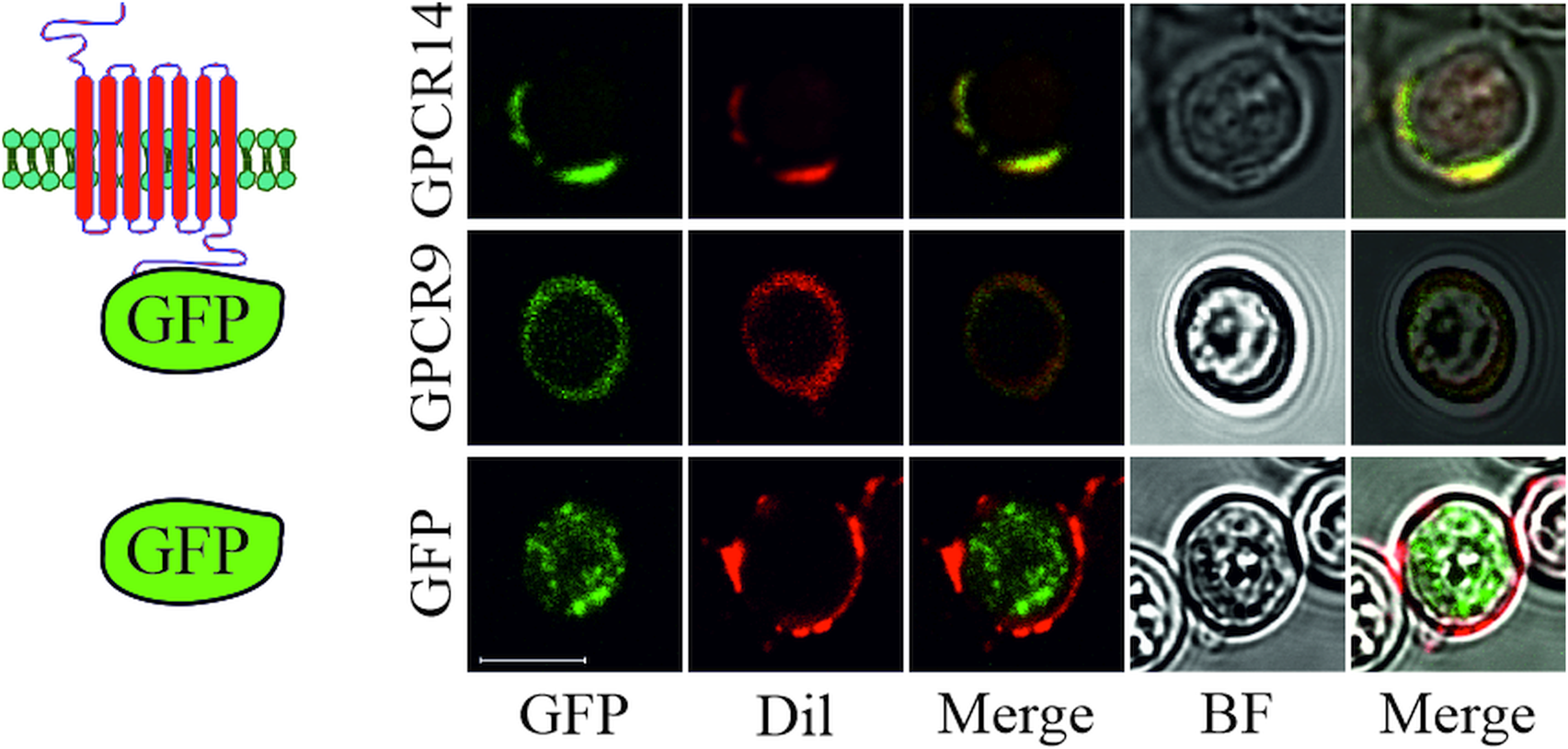
Fig. 3. Subcellular localization of S. mansoni GPCRs in S. cerevisiae. Plasmids coding for eGFP or GPCRs (Smp_203500 (GPCR14) and Smp_244240opt (GPCR9)) tagged with eGFP at the C-terminus were transformed into yeast strain Y178. Fluorescence of eGFP and the membrane-stain Dil were recorded by CLSM. Fluorescent images were merged individually or with bright field (BF) images, respectively. Scale bar, a representative for all pictures: 5 μm.
Monitoring S. mansoni GPCR and neuropeptide interactions
Previous work described the establishment of the MALAR-Y2H system to detect PPIs between secreted and transmembrane proteins (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). The ligand CXCL12 and the mouse receptor CXCR4, representing a well-studied chemokine-signalling pathway, served as model interacting partners in this study.
We employed this system to start deorphanizing S. mansoni GPCRs and therefore generated a library of 47 neuropeptides (Supplementary Table 3) as chimeric proteins fused via a flexible linker to the C-terminal part of the split ubiquitin system. In analogy, GPCRs were cloned as fusion proteins with the N-terminal part of ubiquitin. Mated clones were selected on SD/Trp− Leu− plates to ensure co-expression of bait and prey in the cells (Fig. 1). The combination of receptor GPCR14 and NPP2a revealed growth on SD/Trp− Leu− His− Ade− agar plates. Only weak growth was monitored for all other 46 combinations (exemplified by NPP2b). This suggested a specific interaction of GPCR14 with NPP2a (Fig. 4A). Growth on SD/Trp− Leu− plates showed only slight differences between all combinations indicating that clonal effects were not responsible for our finding. The positive control (CXCR4/CXCL12) showed growth comparable to GPCR14/NPP2a and the negative control (OST1/CXCL12) to all other tested ligands supporting the functionality of the assay. In addition, we investigated the β-Gal activity of the hybrid cells. The results were consistent with the growth assays. Combinations of GPCR14/NPP2a and CXCR4/CXCL12 revealed Miller units of 2.68 and 3.07, respectively (Fig. 4B). Yeast strain AH109 transfected only with bait plasmids NPP2a and NPP2b showed Miller units comparable with the wild-type strain indicating no self-activation or a false positive release of the GAL4 transcription factor. The transcript levels of gpcr14 and npp2 have been shown to be highest in males and unpaired females supporting the possibility of genuine interactors (Supplementary Fig. 3).
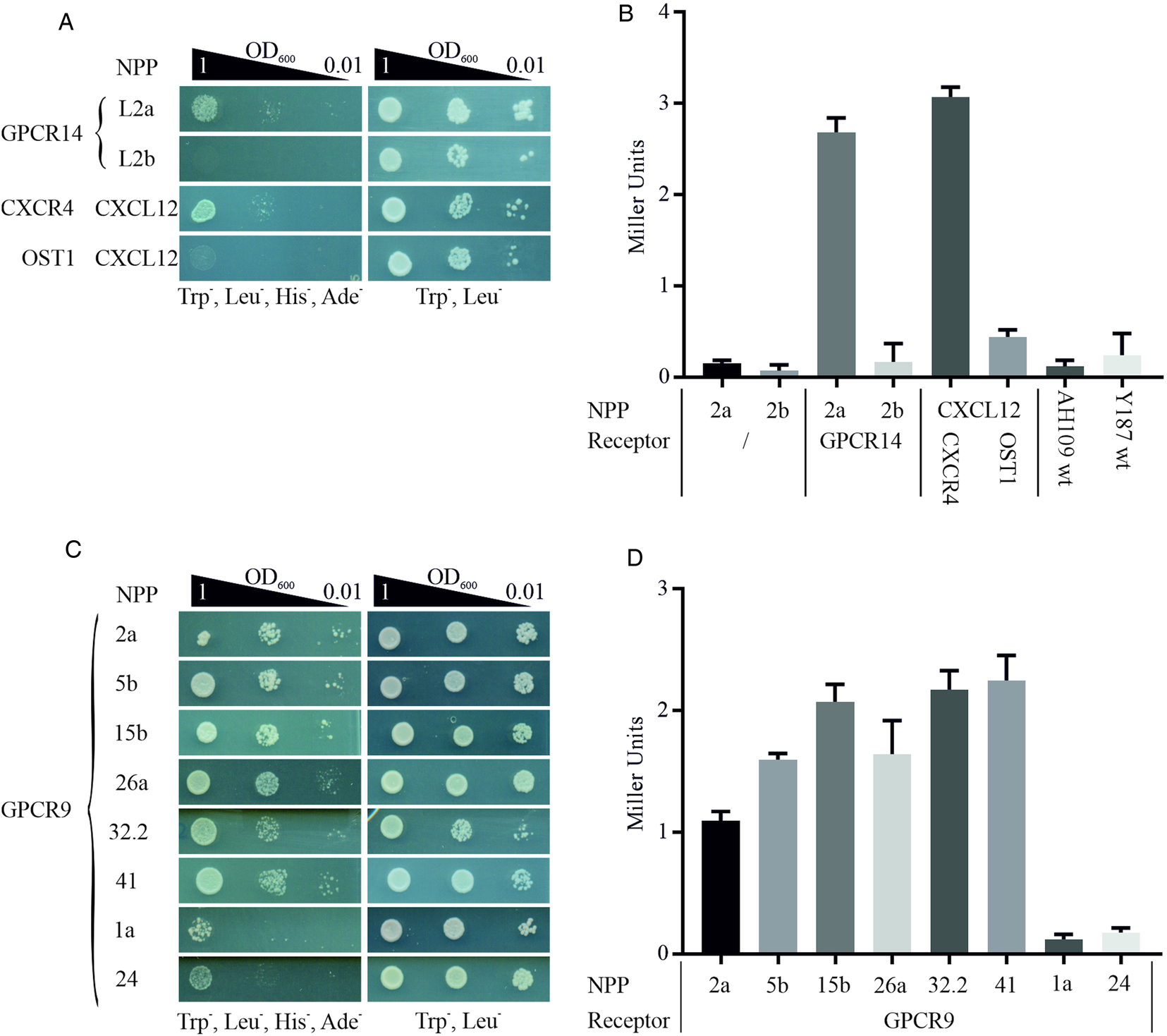
Fig. 4. Detection of protein–protein interactions between GPCRs and hormone peptides. Shown are cell growth assays of yeast strain AH109 transfected with plasmids expressing ligand fusion proteins (NPP), which was mated with yeast strain Y187 transfected with plasmids expressing (A) Smp_203500 (GPCR14) and (C) Smp_244240opt (GPCR9), respectively. The classic chemokine CXCL12 and its known receptor CXCR4 were employed as a positive control (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). OST1 is a transmembrane protein of S. cerevisiae and in combination with CXCL12 used as the negative control. Three different OD600 concentrations of diploid yeast cells were dropped onto SD/Trp− Leu− His− Ade− and Trp− Leu− plates; the latter served as growth control. Colony growth was monitored after 72 and 48 h, respectively. (B, D) ONPG-assays to measure β-Gal activity of diploid cells as in A and C. Cells were lysed in liquid nitrogen and incubated with ONPG for ~30 min at 30°C. OD405 was measured, and Miller Units were calculated (Miller Units = 1000 × OD405/t(min) × OD600). Shown are the mean values of two clones (n = 2).
With GPCR9 as prey, we observed the strongest growth with ligands NPP2a, 5b, 15b, 26a, 32.2 and 41 on SD/Trp− Leu− His− Ade− agar plates (Fig. 4C). The weakest growth was detected for ligands NPP1a and NPP24. The remaining 39 hybrids showed a weak to moderate growth in the range of OD600 1 and 0.1 (data not shown). All tested clones showed equal growth on SD/Trp− Leu− agar plates. The results of the β-Gal assays were in line with the growth assays (Fig. 4D). Miller Units ranged from 1.09 to 2.25 for strongly growing hybrids, and 0.12 and 0.17 for GPCR9/NPP1a and GPCR9/NPP24, respectively. RNA transcripts of gpcr9 have been solely detected in testis of males (Supplementary Fig. 3). Transcript levels of npp5, 2 and 41 were highest in males and unpaired females, whereas npp15, 26 and 32.2 were most prominent in unpaired females. Furthermore, we conducted the MALAR-Y2H assay with the ligand library and GPCR13. In contrast to GPCR9 and GPCR14, we only monitored weak to no growth on SD/Trp− Leu− His− Ade− agar plates with all 47 hybrids (Supplementary Fig. 4).
Discussion
Our current knowledge about PPI networks has been largely derived from Y2H analysis (Suter et al., Reference Suter, Kittanakom and Stagljar2008). The original method was developed to investigate PPI of cytoplasmic proteins (Fields and Song, Reference Fields1989). This groundwork paved avenues for more sophisticated applications to detect PPIs between cytoplasmic proteins and membrane proteins, and between two membrane-associated proteins (Stagljar et al., Reference Stagljar, Korostensky and Johnsson1998). A further modification, MALAR-Y2H system, even allowed the detection of PPIs on the outer cytoplasmic membrane between secreted cytokines and transmembrane receptors (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). We have employed the MALAR-Y2H system to identify PPIs of GPCRs with their putative neuropeptides. Although yeasts are eukaryotic organisms, possessing the necessary machinery for post-translational processing, it is not self-evident that genes from S. mansoni are processed correctly in the heterologous yeast system. Indeed, previous experiments with S. mansoni α-integrins (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015) and findings from this study showed that sequence structure influences transcription. One out of seven tested CDS of S. mansoni GPCRs was not expressed as a full-length transcript. As in the previous study (Parker-Manuel et al., Reference Parker-Manuel, Grevelding and Gelmedin2015), this correlated with a DSM-score of >20, which indicates a recognition sequence of the 3′ mRNA processing machinery of yeast (Graber et al., Reference Graber, McAllister and Smith2002) interfering with the transcription of this GPCR-coding gene. As DSM output, a score indicated the probability of a 3′-processing site being present and activated at that particular cleavage point within this sequence. To overcome this limitation, we optimized the codon sequences without disturbing the aa sequence. Subsequently, we succeeded in expressing the optimized version of Smp_244240opt (GPCR9). Using yeast as a model for gene heterologous gene expression could also affect molecular work with other parasites with a high AT content such as Plasmodium sp. (Gardner et al., Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya, Martin, Fairlamb, Fraunholz, Roos, Ralph, McFadden, Cummings, Subramanian, Mungall, Venter, Carucci, Hoffman, Newbold, Davis, Fraser and Barrell2002; Videvall, Reference Videvall2018).
Using the MALAR-Y2H system, we assumed that schistosome neuropeptides and receptors are transported and integrated into the plasma membrane. This is facilitated by a secretory signal peptide (SP) located at the N-terminus of the fusion protein. This SP usually contains an N-terminal basic aa followed by a stretch containing hydrophobic residues (Heijne, Reference Heijne1990). During protein maturation, the SP is cleaved off. We used the SPs of yeast WBP1 and OST1 for neuropeptides and GPCRs, respectively. It has been demonstrated before that these SPs are feasible to target receptors and ligands to the membrane (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016). Additionally, a prediction tool supported the presence of signal peptides and their cleavage sites in all chimeric proteins used in our study (Almagro Armenteros et al., Reference Almagro Armenteros, Tsirigos, Sønderby, Petersen, Winther, Brunak, Heijne and Nielsen2019). In this pilot approach, we investigated the localization of ligands and GPCRs. As assumed, we observed that ligand fusion proteins such as the representative neuropeptide (L5a) that had been fused to the transmembrane domain of WBP, was targeted to the outer membrane. This corresponds to previous data with the chemokine ligand CXCL12 using the same system (Li et al., Reference Li, Gao, Han, Zhang, Guan, Zhou, Yu and Han2016) confirming the suitability of this approach also for schistosome neuropeptides. For GPCRs, we observed that GPCR9 and GPCR14 localized to the plasma membrane. This was confirmed by a ring-shaped GFP signal, indicating membrane localization and co-localization with a membrane-specific dye. Unexpectedly, the fluorescence signal for GPCR13 was mainly localized in the cytoplasm. In this case, the failure of membrane localization could have different reasons. First, we observed a lack of growth of GPCR13 in combination with all tested neuropeptides. Thus the combination of the chosen SP and the receptor may not have matched and recognized by yeast cells leading to failure of membrane trafficking. To improve GPCR13 membrane trafficking, combinations with different SPs could be tested. Recently, synthetic SPs were developed that enable efficient secretory protein production in yeast that could help in this respect (Yarimizu et al., Reference Yarimizu, Nakamura, Hoshida and Akada2015). Second, some receptors have been reported to be poorly expressed because they get stuck in the membrane of the endoplasmic reticulum, or they are directed to the vacuoles undergoing proteolytic degradation (Reiländer, Reference Reiländer and Weiss1998). Finally, although it appears unlikely to us, we cannot finally exclude that GPCR prediction by bioinformatics (Berriman et al., Reference Berriman, Haas, LoVerde, Wilson, Dillon, Cerqueira, Mashiyama, Al-Lazikani, Andrade, Ashton, Aslett, Bartholomeu, Blandin, Caffrey, Coghlan, Coulson, Day, Delcher, DeMarco, Djikeng, Eyre, Gamble, Ghedin, Gu, Hertz-Fowler, Hirai, Hirai, Houston, Ivens, Johnston, Lacerda, Macedo, McVeigh, Ning, Oliveira, Overington, Parkhill, Pertea, Pierce, Protasio, Quail, Rajandream, Rogers, Sajid, Salzberg, Stanke, Tivey, White, Williams, Wortman, Wu, Zamanian, Zerlotini, Fraser-Liggett, Barrell and El-Sayed2009; Hahnel et al., Reference Hahnel, Wheeler, Lu, Wangwiwatsin, McVeigh, Maule, Berriman, Day, Ribeiro and Grevelding2018) may have resulted in few false-positive results.
Using the MALAR-Y2H system as a novel approach to deorphanize schistosome GPCRs investigating neuropeptide interactions led to first promising results. An obvious advantage of this system is that GPCRs and ligands are co-expressed at the membrane which favours physical proximity as a prerequisite for interaction. This is especially the case for longer neuropeptides, if tertiary structure formation is required for proper interaction (Keire et al., Reference Keire, Bowers, Solomon and Reeve2002). Here, the addition of a linker may be beneficial allowing a higher spatial degree of freedom. In addition, the receptor does not have to be ‘activated’ in the sense of conformational change and G-protein recruitment. A higher degree of spatial freedom may also explain why more than one interaction was found in the case of GPCR9. Although it is well established that GPCRs can interact with more than one specific ligand and vice versa (Luttrell, Reference Luttrell2008), we cannot completely rule out that not all detected neuropeptides are genuine ligands also in vivo. In the heterologous system, ligands are faced with GPCRs, and they may interact based on structural prerequisites. However, it may also be possible that they may not face each other in vivo due to expression in e.g. different tissues or at different time points during development. In addition, the neuropeptides do not undergo a C-terminal amidation, which might be a source of undetected interactions. On the other hand, we detected only one potential ligand (NPP2a) for GPCR14. This finding proves that the system can be selective and leakiness appears to be low since yeast cells only transfected with bait plasmids (NPP2a & NPP2b) showed low Miller Units comparable to the wild type yeast strain AH109. The latter indicated that the C-terminal part of ubiquitin alone is not cleaved by UBP, which prevents false-positive results. In conclusion, the PPI of receptor and ligand was the driving force to ultimately release GAL4 for activating reporter genes.
To substantiate indicative findings of PPIs identified by MALAR-Y2H system, further approaches are needed. One possibility to identify natural ligands of GPCRs is to take advantage of the promiscuous nature of the G-protein subunit Gα 16 that couples a wide range of GPCRs to the phospholipase C-β pathway. Co-expression of Gα 16 along with GPCRs of interest and mitochondrially targeted apoaequorin in CHO cells enable sensitive monitoring of intracellular calcium responses to exogenous ligands. This way the planarian neuropeptide NPY8 has been shown to be the cognate ligand for NPYR1 (Saberi et al., Reference Saberi, Jamal, Beets, Schoofs and Newmark2016). NPY8 plays a crucial role in sexual maturation and germ cell differentiation. Another approach is the use of the yeast pheromone signalling pathway for GPCR-ligand screening (Klein et al., Reference Klein, Paul, Sauvé, Schmidt, Arcangeli, Ransom, Trueheart, Manfredi, Broach and Murphy1998; Minic et al., Reference Minic, Persuy, Godel, Aioun, Connerton, Salesse and Pajot-Augy2005). This requires the development of yeast strains containing a chimeric Gα subunit composed of segments that couple to mammalian GPCRs and yeast segments that interact with the Gβ/Gγ subunits. Hence, an outer signal can be coupled to the pheromone response signalling cascade activating an exogenous reporter gene (in most cases complementing his3 auxotrophs). These procedures have two potential benefits compared to the MALAR-Y2H system. These are free-floating neuropeptides including the possibility for a C-terminal amidation. It has been demonstrated that this modification is essential for biologic activity (Eipper et al., Reference Eipper, Stoffers and Mains1992; McVeigh et al., Reference McVeigh, Mair, Atkinson, Ladurner, Zamanian, Novozhilova, Marks, Day and Maule2011). Furthermore, activation of the receptor by its natural ligand can be monitored in a dose-dependent manner. A S. mansoni G protein-coupled acetylcholine receptor expressed in yeast cells showed enhanced activity when incubated with increased acetylcholine concentrations (100 nM–10 μM) (MacDonald et al., Reference MacDonald, Kimber, Day and Ribeiro2015). Nevertheless, the coupling efficiency of heterologous GPCRs with chimeric Gα subunits is a major issue when the functional response of a receptor is assessed in vivo. In order to use yeast for S. mansoni GPCR functional characterization, there could be a need for yeast genetic modifications to introduce a proper link between the heterologous receptor and the yeast signalling cascade. So far there are yeast strains available consisting of mammalian chimeric Gα subunits. In future experiments, we intend to analyse the obtained PPI in these systems as well to further strengthen our above findings. Indeed, a S. mansoni dopamine receptor was shown to be responsive to dopamine in a dose-dependent manner, whereas other structurally related amines had no effect (Taman and Ribeiro, Reference Taman and Ribeiro2009). The receptor activity was highest with a chimeric Gα gene in which the last five amino acids of yeast Gα were replaced with those of human Gα S. Lower or no receptor activity was detected with strains carrying chimeras of yeast Gα and human Gα i2, Gα 12, Gα 0 and Gα q. This supports the conclusion that the proper linking of GPCRs to the genetically modified pheromone response pathway has to be carefully tested.
In summary, the data of our pilot study suggest that the MALAR-Y2H system provides a suitable way to deorphanize GPCRs of interest and to obtain first hints of their putative ligands.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182019001756
Acknowledgements
We thank Martin Haimann, Christina Scheld and Georgette Stovall for excellent technical support.
Financial support
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), grant GR1549/7-3 (CGG), and the Wellcome Trust, grant 107475/Z/15/Z (CGG; FUGI) with respect to the transcriptomics data.
Conflict of interest
None.







 Open access
Open access