


Obesity is often accompanied by a state of chronic subclinical inflammation characterized by an increase in the production of pro-inflammatory cytokines such as IL-6, plasminogen activator inhibitor-1, monocyte chemoattractant protein-1 and TNF-α, as well as a change in endocrine and metabolic adipose tissue function. An important step in this development is considered to be the crosstalk between adipocytes and infiltrating macrophages, but the full chain of events leading to subclinical inflammation in obese individuals is not yet fully understood(Reference Hoppmann, Perwitz and Meier1). In obesity, adipocytes release cytokines into the circulation, which stimulate hepatic C-reactive protein (CRP) production and a clear positive association between BMI (kg/m2) and CRP has been observed(Reference Zimmermann, Anty and Tordjman2, Reference Wang and Nakayama3). Hence, for scientific purposes, CRP is commonly used as a marker of obesity-related low-grade inflammation. However, studies have revealed individual differences in CRP levels which cannot be explained by location and degree of fatness alone, and the causes of these differences are poorly understood(Reference Kelley-Hedgepeth, Lloyd-Jones and Colvin4–Reference Florez, Castillo-Florez and Mendez6). In line with this, healthy individuals with a BMI within normal limits, but with a high body fat percentage, may show signs of systemic subclinical inflammation(Reference De Lorenzo, Del Gobbo and Premrov7).
There are several closely linked hypotheses seeking to explain obesity-related low-grade inflammation, including necrotic adipocyte death, endoplasmic reticulum (ER) stress and lipotoxicity.
According to Cinti et al. (Reference Cinti, Mitchell and Barbatelli8) the adipocyte, when overloaded with TAG, is not capable of undergoing normal apoptosis, but instead undergoes a necrotic-like cell death accompanied by inflammation. The release of lipid droplets and other components from the dead adipocyte may then act as a trigger for macrophage infiltration(Reference Cinti, Mitchell and Barbatelli8). The activated macrophages complement obesity-associated adipocyte death by releasing cytotoxins such as TNF-α, nitric oxide and reactive oxygen species, hence leading to low-grade inflammation.
In addition to this hypothesis, recent work by Hotamisligil's group(Reference Özcan, Cao and Yilmaz9) indicates that the ER also plays an important role in the initiation of obesity-related inflammation. The consequence of increased lipid storage in the adipocyte is dysfunction of the ER, leaving it unable to properly fold proteins. This type of cellular stress leads the adipocyte to reduce secretion of adiponectin and to increase secretion of cytokines such as TNF-α, explaining the very beginning of the inflammatory process(Reference Özcan, Cao and Yilmaz9). The secretion of TNF-α triggers a cascade of unfortunate events starting with either preadipocytes or endothelial cells producing monocyte chemoattractant protein-1, hence attracting macrophages to the adipose tissue(Reference Wellen and Hotamisligil10). Also, increased secretion of leptin and/or decreased production of adiponectin may enhance macrophage infiltration and hence lead to low-grade inflammation.
The lipotoxicity hypothesis relates to the assumption that TAG are biologically inert, whereas NEFA accumulating in non-adipose tissue are harmful. This is explained by the fact that adipocytes, in times of energy abundance, store the extra energy in the form of TAG and produce leptin, which in turn limits energy intake by influencing the hypothalamus and stimulating oxidation of fatty acids in the non-adipose tissue. However, in the case of continuous energy excess, these mechanisms are assumed to fail, causing fatty acids to accumulate in the non-adipose tissue such as liver, skeletal muscle, heart muscles, pancreatic islets and the perivascular tissue, and thus leading to ER stress and necrotic cell death, possibly caused by the harmful lipid metabolites such as diacylglycerol and ceramide. This leads to dysfunction of the organs and inflammation(Reference Unger, Clark and Scherer11–Reference Eckardt, Taube and Eckel13). These closely intertwined hypotheses all result in a release of inflammatory cytokines from the adipose tissue, which again causes production of acute-phase proteins from the liver, most notably CRP.
Despite detailed hypotheses of the mechanisms underlying obesity-related inflammation, the environmental and genetic factors triggering such mechanisms remain unclear. Identification of determinants for adipose tissue inflammation is a crucial step in enabling targeted actions against obesity-related inflammation and thereby to prevent associated clinical conditions such as type II diabetes, CVD and even cancer.
A wide range of genetic and environmental factors may be considered to play a role in the aetiology of obesity-related inflammation and may consequently also explain the inter-individual variation in inflammation among obese individuals (Fig. 1). Infections are known to cause an increase in inflammatory markers and potentially also play a role in the development of obesity. Likewise, the gut microbiota as well as the intake of specific nutrients may play a role in this regard, since a considerable part of the human immune system is located in the intestinal mucosa and responds to luminal antigens (i.e. both nutrients and pathogens). Furthermore, physical activity may affect both the risk of obesity and systemic levels of inflammatory markers and may therefore also influence the risk of developing obesity with inflammation. Lastly, specific genes may be hypothesized to be involved in the regulation of both obesity and inflammation. Altogether, a quantitative assessment of environmental and genetic determinants of obesity-related inflammation would require an analytical-epidemiological approach.
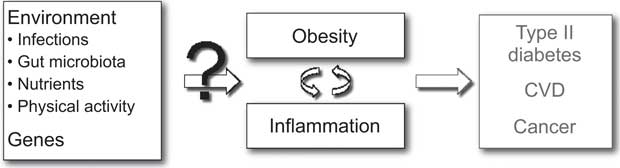
Fig. 1 Potential determinants of inter-individual differences in inflammation related to obesity
The aims of the present study were: (i) to assess current knowledge on the potential epidemiological determinants for development of adipose tissue inflammation mentioned above; and (ii) to set the scene for further research within this area.
Methods
In the current narrative review we identified literature on potential determinants of both obesity and inflammation by searching MEDLINE (1966–2010), applying the MeSH terms ‘obesity’ and/or ‘inflammation’ combined with a free text search on the terms ‘infection’, ‘bacteria’, ‘virus’, ‘gut microbiota’, ‘nutrition’, ‘nutrients’, ‘food’, ‘physical activity’, ‘genes’ and ‘genome wide association studies’. We started out with the specific criterion that only studies reporting on our exposures of interest in combination with both inflammation and body weight as outcomes should be included, but during the search process we had to broaden the inclusion criteria to cover also articles reporting on either inflammation or body weight in combination with the exposures of interest. Reference lists of included papers were assessed to disclose additional literature on the topic.
Results: determinants of adipose tissue inflammation
Infections
Obesity has spread globally in a way that, epidemiologically, shows remarkable comparison to that of an infectious disease. Infections are known to affect the level of local and/or systemic inflammation, and studies in human populations further suggest a role for human Adenovirus-36 (Ad-36), SMAM-1 virus, Helicobacter pylori and Chlamydia pneumoniae in the development of obesity.
Ad-36 is an antigenically unique virus, which shows little similarity with other human adenoviruses(Reference Dhurandhar14). The transmission of the virus occurs via respiratory, faeco-oral and sexual pathways, and also through contaminated personal objects(Reference Suplicy Hde and Bornschein15). An American study of 502 individuals observed Ad-36 antibodies in 30 % of obese v. 11 % of lean individuals(Reference Atkinson16). Likewise, a twin study found that twenty-six of eighty-nine twin pairs were discordant for Ad-36 antibodies, which related to significant differences in BMI and body fat percentage(Reference Dhurandhar14). A study of 540 Korean adults concluded Ad-36 to be strongly associated with overweight but not obesity(Reference Na, Kim and Lee17). Interestingly, an increased production of IL-6 in adipocytes affected with Ad-36 has also been observed(Reference Bouwman, Visseren and Bouter18).
SMAM-1 is an avian adenovirus, known to infect chickens and cause excessive fat accumulation in these(Reference Dhurandhar14). SMAM-1 virus has also been associated with human adiposity, with significantly higher BMI being observed in antibody-positive compared with antibody-negative individuals(Reference Dhurandhar, Kulkarni and Ajinkya19). Likewise, it has been suggested that infections with H. pylori and C. pneumoniae are associated with increased BMI(Reference Thjodleifson, Olafsson and Gislason20). However, the concomitant role of these pathogens in the development of obesity-related inflammation was not studied.
In animals, Ad-5, Ad-37, Canine Distemper Virus (CDV), Rous-Associated Virus Type-7 (RAV-7), Borna Disease Virus (BDV) and Scrapie-Agent(Reference Dhurandhar14–Reference Atkinson16, Reference Atkinson, Dhurandhar and Allison21–Reference Carp, Kim and Callahan35) have been associated with obesity, but studies did not encompass the aspect of concomitant inflammation.
Gut microbiota
Trillions of bacteria reside in the human gastrointestinal tract, with at least 500 to 1000 distinct bacterial species, collectively known as the gut microbiota(Reference DiBaise, Zhang and Crowell36–Reference Frank, St Amand and Feldman40).
The constitution of the gut microbiota has been suggested to play a role in both systemic inflammation and development of obesity by affecting nutrient acquisition and energy regulation. Accordingly, obese and lean people may have differences in their gut microbiota(Reference DiBaise, Zhang and Crowell36).
The gut microbiota has primarily been studied in animals, showing that young conventionally reared mice have 40 % higher body fat content than their gnotobiotic (germ-free) counterparts(Reference Bäckhed, Ding and Wang41). The latter, in turn, increase their body fat by up to 60 % after exposure to transplanted intestinal flora from the conventionally reared mice with concomitant adipocyte hypertrophy, insulin resistance, and increased levels of glucose and leptin(Reference Bäckhed, Ding and Wang41). Germ-free mice receiving a diet rich in sugar and fat appear to be protected from diet-induced obesity via elevated levels of fasting-induced adipocyte factor and through increased levels of phosphorylated AMP-activated protein kinase(Reference Bäckhed, Manchester and Semenkovich42). Furthermore, characterization of the distal gut microbiome in genetically obese leptin-deficient (ob/ob) mice v. lean (ob/+ and +/+) mice has shown that the ob/ob mice contain genes expressing enzymes capable of breaking down polysaccharides in the form of resistant starch and poorly digested carbohydrates to SCFA such as acetate, propionate and butyrate. These polysaccharides are indigestible to their lean counterparts and therefore the stools of the obese mice contain less energy and more end-products of fermentation(Reference Turnbaugh, Ley and Mahowald43, Reference Topping and Clifton44). This led to the idea that the extraction of additional energy from ingested food was facilitated by the gut microbiota of the ob/ob mice. Lean gnotobiotic mice transplanted with gut microbiota from the ob/ob mice also gain substantially more weight than recipients of ob/+ and +/+ microbiota, which further supports the idea of a microbial component in the pathogenesis of obesity. More specifically, obese mice have fewer Bacteroidetes and more Firmicutes in their caecal microbiota than their lean counterparts(Reference Ley, Bäckhed and Turnbaugh45).
Regarding inflammation, recent evidence suggests that normal gut microbiota protects against inflammatory diseases(Reference Maslowski, Vieira and Ng46). In germ-free mice, induced colitis results in significantly worse colonic inflammation than in conventionally reared mice, which decreases after colonization with flora from the conventionally reared mice(Reference Maslowski, Vieira and Ng46). Feeding mice a high-fat diet changes the composition of the gut microbiota from being predominantly Gram-positive to being predominantly Gram-negative and leads to increased endotoxaemia(Reference Cani, Amar and Iglesias47). Chronic metabolic endotoxaemia may induce obesity, insulin resistance and type II diabetes, and lipopolysaccharide derived from Gram-negative bacteria may be a key factor connecting the gut microbiota to inflammation induced by a high-fat diet(Reference Cani, Amar and Iglesias47).
In human studies, obese individuals randomly assigned to a fat- or carbohydrate-restricted low-energy diet increase their number of Bacteroidetes and decrease their percentage of Firmicutes along with weight loss(Reference Ley, Turnbaugh and Klein48). Likewise, among twins, obesity correlates with a lower number of Bacteroidetes and lower diversity than among lean twin siblings(Reference Turnbaugh, Hamady and Yatsunenko49). However, the concomitant effect on inflammation has not been assessed in human studies, as was done in the animal study described above(Reference Cani, Amar and Iglesias47).
Nutrients and physical activity
Nutrients
Recently, Summerbell et al.(Reference Summerbell, Douthwaite and Whittaker50) made a comprehensive systematic review on the association between nutrients, physical activity and obesity. Only consumption of fast foods, intake of non-caloric sweeteners and breast-feeding were found to be directly associated with excessive weight gain and obesity. Four studies on the association between fast food and obesity were included in the review, all linking frequency of fast-food consumption to higher BMI and weight gain in women(Reference Jeffery and French51–Reference French, Harnack and Jeffery54). Likewise, higher levels of consumption of non-caloric sweeteners were linked to obesity and subsequent excess weight gain(Reference Summerbell, Douthwaite and Whittaker50). Finally, compared with being bottle-fed, being breast-fed was associated with slightly lower levels of weight gain and obesity(Reference Summerbell, Douthwaite and Whittaker50). The epidemiological evidence was found to be substantial and generally consistent(Reference Owen, Martin and Whincup55, Reference Arenz, Rückerl and Koletzko56). The review did not address whether consumption of fast foods, intake of non-caloric sweeteners and breast-feeding influenced inflammation concomitantly with BMI. However, high-fat meals have been associated with an increase in inflammatory markers according to a study by Lundman et al.(Reference Lundman, Boquist and Samnegård57), who showed an increased plasma level of IL-6 in response to postprandial lipidaemia.
Although different subtypes of fat were not shown by Summerbell et al.(Reference Summerbell, Douthwaite and Whittaker50) to cause obesity, other studies suggest that they can be linked to inflammation. Trans-fatty acids (TFA) have shown pro-inflammatory effects according to randomized controlled trials (RCT) and observational studies, where increased intake of TFA has led to increased concentrations of inflammatory markers such as TNF-α and CRP(Reference Micha and Mozaffarian58). Evidence of a concomitant effect of TFA on obesity and inflammation is lacking, which may be due to the fact that very few studies have assessed the impact of TFA on fatness (but rather on cardiovascular risk, which may be linked to both fatness and inflammation). However a recent study by Nielsen et al.(Reference Nielsen, Nielsen and Jakobsen59) suggests that a relatively low contribution (as compared with former RCT and observational studies) of TFA (1·3 g/d) in the diet is not correlated with abdominal fatness or levels of CRP and IL-6.
In contrast to TFA, PUFA may possess anti-inflammatory effects. In an RCT by Micallef and Garg(Reference Micallef and Garg60) it was possible to demonstrate a significant reduction in investigated inflammatory markers after 3 weeks of dietary intervention with n-3 PUFA and plant sterols, the control group receiving sunflower oil. The reduction was most obvious in circulating CRP levels with a 39 % decrease, but also TNF-α, IL-6 and leukotriene B-4 were significantly decreased. Further, we recently demonstrated an inverse relationship between the intake of n-3 PUFA and body fat, but without a concomitant beneficial effect on systemic levels of inflammatory markers.
Resveratrol is another dietary component, found in the skin of red grapes and other fruits, which may play a key role in the inflammatory process through inhibition of cyclooxygenase-2 activity and production of prostaglandins. Animal and in vitro studies suggest that resveratrol also inhibits IL-8 and granulocyte-macrophage colony-stimulating factor and suppresses IL-6 gene expression, all leading to decreased inflammation(Reference Fan, Zhang and Jiang61). At the same time, resveratrol possesses anti-obesity effects though decreased adipogenesis and viability in maturing preadipocytes, and increased lipolysis and reduced lipogenesis in mature adipocytes(Reference Baile, Yang and Rayalam62).
Lastly, in a systematic review by North et al. on dietary fibre and inflammation(Reference North, Venter and Jerling63) six out of seven identified studies found significantly lowered CRP concentrations as a result of increased fibre consumption, with a trend towards larger CRP reduction with increased dietary fibre intake. Still, the role of dietary fibre and its association with obesity is uncertain(Reference Summerbell, Douthwaite and Whittaker50).
Physical activity
In their comprehensive review, Summerbell et al.(Reference Summerbell, Douthwaite and Whittaker50) also included thirty-eight studies on physical activity and obesity. However, only two of eighteen studies in adults and three of twenty studies in children used objective methods (rather than questionnaires) to determine the level of physical activity. The conclusion of the review was that there is no association between physical activity and BMI. Even the five studies using objective methods such as accelerometry and doubly labelled water did not show a consistent association between physical activity and BMI. Still, studies have found an inverse correlation between physical activity and inflammatory markers. Physical activity intervention studies have shown that (compared with receiving only written information on lifestyle intervention) an increase in physical activity can reduce CRP concentrations in obese individuals(Reference Nicklas, Ambrosius and Messier64, Reference Andersson, Boman and Jansson65). Likewise, Lin et al. have recently shown that cardiorespiratory fitness was associated with lower levels of CRP and was, in addition, inversely associated with insulin resistance(Reference Lin, Chen and Kuo66). A recent review by Wärnberg et al.(Reference Wärnberg, Cunningham and Romeo67) concludes that some studies have shown an association between physical activity and inflammatory markers independent of fatness. However, if physical activity and obesity should be inversely related, it remains unclear whether the anti-inflammatory health benefits of a physically active lifestyle are caused by exercise per se or result from favourable changes in body composition.
Genes
In obese individuals, a number of genetic alterations have been identified that are believed to have a partial and additive effect on the development of obesity(Reference Ichihara and Yamada68). This association has been made obvious by studies of adoptees who resemble their biological relatives but not their adoptive relatives and by numerous twin studies, suggesting that genetics accounts for 50–90 % of the variance in BMI(Reference Sørensen, Price and Stunkard69, Reference Stunkard, Sørensen and Hanis70).
The introduction of genome-wide association studies has led to further findings(Reference Loos and Bouchard71). Numerous genes influencing BMI and fat storage have been located (in different organisms), including eleven single gene mutations, 244 knockout or transgenic animal models and 135 different candidate genes associated with obesity-related phenotypes. Variants in more than twenty different human genes associated with obesity-related phenotypes have been confirmed in at least five independent studies, and two genes in particular have been convincingly associated with obesity: FTO (fat mass and obesity associated gene) and MC4R (melanocortin-4 receptor gene)(Reference Andreasen and Andersen72–Reference Qi and Cho74).
FTO is a large gene consisting of nine exons with a length of more than 400 kb, located on human chromosome 16. The relationship to obesity is caused by an SNP located in a cluster of closely situated SNP in the first intron of the gene. This was first discovered by Frayling et al. (Reference Frayling, Timpson and Weedon75) through a genome-wide association study with primary focus on the aetiology of type II diabetes. A highly significant association with type II diabetes was found, but this was entirely due to the effect of FTO on fatness. Since this discovery, results have been replicated in more than ten cohort studies comprising more than 40 000 individuals and FTO is now regarded as a risk factor for common obesity, although it explains only a small fraction of the overall variation of BMI in the population. It is expressed in various tissues, including adipose tissue, pancreatic islets, skeletal muscle and brain tissue, including the hypothalamic regions, suggesting a role in the regulation of appetite. However, it has yet to be explained how this directly links to obesity(Reference Loos and Bouchard71, Reference Andreasen and Andersen72). Furthermore, a German study(Reference Fisher, Schulze and Stefan76) showed that a common and obesity-related variant of FTO, rs9939609, was significantly associated with increased levels of CRP. These findings were significant, independent of the adiposity level and showed a 1·14-fold increase in circulating CRP concentration for men and 1·12-fold increase for women per risk allele. However, a recent study by Zimmermann et al. in a population of Danish men representing a broad range of BMI did not find the fatness-associated FTO rs9939609 A-allele to have a fatness-independent effect on systemic inflammation(Reference Zimmermann, Skogstrand and Hougaard77).
MC4R codes for a melanocortin receptor and is part of the G-protein coupled receptor family, signalling through activation of adenylyl cyclase, located on chromosome 18. Studies have shown that MC4R is important in the regulation of energy balance, having influence on food intake and energy expenditure. Numerous studies suggest that SNP in the vicinity of the MC4R gene coding sequence are associated with obesity and waist circumference(Reference Ichihara and Yamada68, Reference Chambers, Elliott and Zabaneh78). In addition, Trevaskis et al.(Reference Trevaskis, Gawronska-Kozak and Sutton79) showed that MC4R knockout mice fed a high-fat diet had an increased level of inflammatory markers in adipose tissue and reduced serum adiponectin, which led to severe insulin resistance. This result was compared with MC3R knockout mice that exhibited normal adiponectin levels and a delayed inflammatory response, regardless of diet and obesity.
Another SNP being convincingly associated with obesity is located in the promotor region of the TNF-α gene(Reference Brand, Schorr and Kunz80). Furthermore this variant of the gene has been shown to increase transcriptional activation of TNF-α, which, according to a meta-analysis, led to a 1·25-fold increase in fasting plasma insulin levels, thus indicating a possible link to inflammation(Reference Sookoian, González and Pirola81). Lastly, SNP in the leptin receptor gene (LEPR) have been found to be related both to obesity and plasma CRP levels(Reference Bochud, Marquant and Marques-Vidal82–Reference Zhang, Gottardo and Mlynarski84).
Discussion
It is well established that obesity is associated with a state of low-grade inflammation, but the determinants of the initiation of obesity-related inflammation remain uncertain, hence leaving us unable to explain inter-individual differences in levels of inflammation among obese individuals. We suggest a potential role for Ad-36, the gut microbiota, TFA, and the four genes FTO, MC4R, TNF-α and LEPR in the causation of obesity with inflammation, but also disclose a surprising lack of literature focusing on the role of environmental or genetic factors in the development of obesity with or without inflammation (Table 1).
Table 1 Determinants of obesity, inflammation and obesity with inflammation, according to current evidence
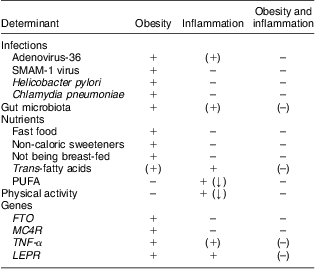
FTO, fat mass and obesity associated gene; MC4R, melanocortin-4 receptor gene; LEPR, leptin receptor gene.
+ indicates a reported association; (+) indicates a suggested association; – indicates no known association; (–) indicates partly lack of evidence of an association; (↓) indicates that the reported association is negative (i.e. the determinant decreases the risk of obesity or inflammation).
Pathogens may be hypothesized to influence the development of obesity with inflammation, since an association between obesity per se and respectively Ad-36, SMAM-1, H. pylori and C. pneumoniae has been observed in human populations. In animals, infectious agents such as CDV, RAV-7, BDV, Scrapie-Agent, Ad-37 and Ad-5 have also been reported to be adipogenic. However, despite a known effect of pathogens on inflammatory response, only one study specifically assessed the influence of an adipogenic pathogen on inflammation, suggesting that Ad-36 increases the production of IL-6(Reference Thjodleifson, Olafsson and Gislason20). In line with this, one may hypothesize that pathogens directly affecting the adipose tissue (such as Ad-36 and SMAM-1) are more likely to cause adipose tissue inflammation than obesity-related pathogens affecting, for example, the brain (BDV and CDV). Hence, studies investigating the effect of pathogens on obesity combined with local and systemic inflammation are needed, and may for ethical reasons rely on an epidemiological approach using antibody assessments in well-characterized longitudinal cohort studies with repeated blood tests (for assessment of both antibodies and inflammation), repeated anthropometric measurements and (if possible) tissue samples to determine local concentration of specific antibodies as well as local inflammation.
The gut microbiota is another perspective of potential microbial influence on human obesity and inflammation. In contrast to infections with external pathogens, the microbiota persists and is considered fairly stable over time. The establishment of the gut microbiota in early life makes it of particular interest. In Denmark, there has been a birth cohort-associated increase in obesity during two time periods (1940–1955 and 1970 onwards)(Reference Olsen, Baker and Holst85) and individuals contributing to this increase in incidence were found to be obese from early childhood, hence suggesting that environmental factors present in the first years of life are essential in development of obesity. A possible interplay between the gut microbiota and obesity is supported by the observed association between the composition of the microbiota and human BMI(Reference Ley, Turnbaugh and Klein48, Reference Turnbaugh, Hamady and Yatsunenko49) and by the different mechanisms for energy harvest in obese and lean mice(Reference Bäckhed, Manchester and Semenkovich42). Knowing that the gut microbiota also has a protective role in the development of inflammation, it is possible, as advocated by Cani et al.(Reference Cani, Amar and Iglesias47), that the gut microbiota may be a determinant not only for obesity and inflammation separately but also for the two in combination. In order to determine whether the composition of the microbiota influences the inter-individual degree of obesity with inflammation, prospective cohort studies of healthy individuals are needed to assess the association between objectively measured weight changes, changes in the composition of the gut microbiota (assessed via stool samples or samples obtained from colonoscopies) and markers of respectively systemic inflammation, such as levels of CRP, and local inflammation, as assessed in fat tissue biopsies.
In addition to pathogens and the intestinal microbiota, nutrients and physical activity also need further consideration. Summerbell et al.'s(Reference Summerbell, Douthwaite and Whittaker50) extensive review only found fast foods, non-caloric sweeteners and not being breast-fed to be significantly associated with obesity. However, only observational studies with a follow-up period of more than 1 year were included in the review and many studies were based on self-reported data on body weight and food consumption, with a related risk of bias. Most importantly, inflammation was not the area of focus in the review. Still, certain nutrients have been linked to inflammation, among which are TFA. Intake of TFA has an unfortunate influence on the lipid profile and may cause visceral adiposity(Reference Micha and Mozaffarian58), suggesting that high intakes of TFA cause obesity in manners of definition and classification, as well as systemic inflammation. The n-3 PUFA, on the other hand, are known to decrease inflammation in other inflammatory conditions, but a potential protective effect on development of obesity, and especially obesity with inflammation, needs further investigation. The same is the case for intake of dietary fibre, which was found to be anti-inflammatory but not to prevent obesity. Resveratrol is probably the most convincing factor with influence on this condition.
Again, future studies should be prospective of nature and use objective and detailed measures of body fatness and local and systemic inflammation. Furthermore, the use of validated measures of food intake is crucial in this regard, as is adjustment for relevant confounders in relation to food intake and preferably also for the other potential determinants of adipose tissue inflammation, especially physical activity.
Physical activity is often regarded one of the cornerstones in fighting the obesity epidemic, although the association remains debated(Reference Summerbell, Douthwaite and Whittaker50). Reporting bias may influence observations or physical activity may lead to increased muscle mass resulting in an unaltered BMI. On the other hand, numerous studies support an impact of physical activity on level of inflammation. Hence, physical activity may still play a role in preventing and/or reversing obesity-related low-grade systemic inflammation and future studies should address both of these aspects. The reversing capability of physical activity on obesity with systemic inflammation could ideally be assessed in an RCT of obese individuals with baseline anthropometric measures plus measures of local and systemic inflammation and with either comparable or detailed measures of dietary intake during the physical activity intervention. Repeated blood samples and fat tissue biopsies would be needed throughout the study period in order to determine the effects on systemic and local inflammation, which together with changes in anthropometry would serve as endpoints of the study. The preventive role of physical activity on obesity with inflammation may more realistically be assessed in a large population-based study with relevant and repeated measures of the endpoints and confounders of interest.
The role of genes and their association with obesity has been made clearer by genome-wide association studies identifying more than twenty genes convincingly related to obesity, out of which four have also been shown to be related to inflammation. However, it is still believed that an obesity-promoting environment determines whether the obese genotype will result in the obese phenotype(Reference Sørensen, Price and Stunkard69, Reference Stunkard, Sørensen and Hanis70). Therefore it can be speculated that SNP causing a genotype associated with inflammation will also need inflammatory-promoting environmental factors to result in the obesity–inflammatory phenotype. However, such gene–environment interactions in the development of adipose tissue inflammation remain to be investigated. It is possible that the genetic influence on obesity and inflammation may – at least partially – explain the inter-individual differences in this condition, and environmental triggers would then be essential targets for intervention. Ideally, all potential environmental and genetic determinants for the development of obesity with inflammation should be assessed in a large-scale epidemiological study, but the realistic approach to this essential scientific question is probably a step-wise process, assessing the suggested determinants one by one, as suggested in the paragraphs above. In a second step, factors that in the first are found to be associated with obesity and concomitant inflammation should be assessed together, in order to determine the individual effect of each factor per se.
Some potential limitations of the present review need consideration. While revealing a still largely unexplored research area of potential environmental and genetic factors playing a role in inter-individual differences in level of obesity-related inflammation, we cannot claim to present an exhaustive but rather a suggestive list of determinants, which represent candidate areas to explore further. Also, the lack of a definition of, or at least a unique measure of, obesity-related inflammation is a limitation for the conduct and comparison of both existing and future studies. Local inflammation in the adipose tissue is probably the most straightforward way to define obesity-related inflammation, but the problem with this definition – from an epidemiological perspective – is the lack of easily accessible biomarkers reflecting the specific type and level of local inflammation. Hence, most studies assessing the role of obesity-related inflammation in development of type II diabetes, CVD and cancer are using CRP as a marker of inflammation. However, it remains uncertain whether CRP is the optimal measure of obesity-related inflammation and if so, at what level CRP can be defined as subclinically increased. The use of fat tissue samples could be a way to investigate how the CRP level is affected by inflammation in adipose tissue, hence elucidating the value of CRP as a marker of obesity-related inflammation. However, despite the apparent lack of a better definition of the obesity–inflammatory phenotype, obesity-related inflammation may only become clinically important when it acts systemically, and then CRP may actually turn out to be the best measure available from an epidemiological perspective. This assumption, however, also needs to be addressed.
Conclusions
After assessing numerous potential determinants of the development of obesity with inflammation, the primary conclusion of the present literature review is a surprising lack of literature on the subject and, hence, a largely unexplored research area.
Among identified environmental factors potentially explaining inter-individual differences in obesity-related inflammation are Ad-36, the gut microbiota and high intake of TFA. In addition, four genes (FTO, MC4R, TNF-α and LEPR) are suggested to be associated with both obesity and inflammation, but studies were few. Once a specific definition of the inflammation–obesity phenotype has been agreed upon, a broader clinical- and genetic-epidemiological understanding of the aetiological role of genes and environmental factors in development of adipose tissue inflammation is needed. This is required not least considering the huge impact of this condition on public health worldwide and the need for identification of determinants which can be targeted.
Acknowledgements
The study was financed through a Female Research Leader grant (no. 09-066323) from the Danish Council of Independent Research to T.J. Also, this work was carried out as a part of the research programme of the Danish Obesity Research Centre (DanORC; www.danorc.dk), granted by the Danish Council for Strategic Research (Grant 2101-06-0005). The authors declare to have no conflicts of interest in relation to the present manuscript. B.J.v.S. and E.N.A. hold joint first authorship. The study was initiated by T.J. B.J.v.S. and E.N.A. performed the literature search and drafted the manuscript under the supervision of T.J. The manuscript was critically revised by T.I.A.S and T.J. All authors approved the final version of the manuscript.


