


Endocannabinoids are endogenously produced compounds that can target cannabinoid CB1 and/or CB2 receptors, both of which are G protein-coupled( Reference Pertwee, Howlett and Abood 1 ). The most investigated of these endocannabinoids have been arachidonoylethanolamide (anandamide) and 2-arachidonoyl glycerol, each of which can activate both CB1 and CB2 receptors( Reference Pertwee, Howlett and Abood 1 ). Endocannabinoids and cannabinoid CB1 and CB2 receptors, together with the processes responsible for endocannabinoid biosynthesis, cellular uptake and metabolism, constitute the ‘endocannabinoid system’( Reference Pertwee 2 ). Importantly, there is convincing evidence, albeit mainly from animal experiments, that there are a number of disorders in which the endocannabinoid system up-regulates in a manner that reduces or abolishes the unwanted effects of these disorders or even slows disease progression. Signs of this ‘autoprotective’ up-regulation include increases in endocannabinoid release, in the expression levels of cannabinoid CB1 or CB2 receptors and/or in the coupling efficiency of these receptors( Reference Pertwee 2 ). This evidence has prompted research directed at seeking out the best strategies for exploiting the production of such autoprotection by the endocannabinoid system in the clinic( Reference Pertwee 2 , Reference Pertwee 3 ). Currently attracting particular attention are a number of ‘indirect’ potential strategies that involve inhibiting the cellular uptake or intracellular metabolism of an endocannabinoid following its endogenous release with the objective of increasing its concentrations at sites in the body at which it is inducing beneficial effects. The remainder of this paper focuses on these ‘indirect’ strategies and their therapeutic potential.
Potential therapeutic applications for inhibitors of endocannabinoid metabolism
For the inhibition of endocannabinoid metabolism, attention has so far focused largely on two enzymes: fatty acid amide hydrolase (FAAH), which is a major metabolising enzyme of anandamide but can also metabolise 2-arachidonoyl glycerol, and monoacylglycerol (MAG) lipase, which is thought to be largely but not exclusively responsible for the metabolism of 2-arachidonoyl glycerol (reviewed in( Reference Pertwee 2 , Reference Di Marzo 10 )). It is noteworthy that there are now known to be at least two forms of FAAH, FAAH-1 and FAAH-2 (which is not expressed by rodents), and also that anandamide and 2-arachidonoyl glycerol can be metabolised to varying extents by a number of enzymes in addition to FAAH or MAG lipase( Reference Alvarez-Jaimes and Palmer 4 , Reference Zhao and Abood 5 , Reference Piscitelli and Di Marzo 49 ). Thus, anandamide can be metabolised by N-acylethanolamine hydrolysing acid amidase, and 2-arachidonoyl glycerol by MAG kinase, α,β-hydrolase (ABHD)6 and ABHD12. Furthermore, both these endocannabinoids are good substrates for cytochrome P450 enzymes, lipoxygenases and cyclooxygenase (COX)-2. MAG lipase, ABHD6 and ABHD12 are all serine hydrolases. However, unlike MAG lipase, which is present on the intracellular surface of cell membranes, these other two hydrolases are both located within the membranes of the cells in which they are expressed, with the active site of ABHD12 facing the outer surface of these membranes and that of ABHD6 located on their inner, cytoplasmic surface( Reference Blankman, Simon and Cravatt 6 ). MAG lipase, ABHD12, ABHD6 and other enzymes (including FAAH) are thought to account, respectively, for about 85, 9, 4 and 2% of total brain-membrane 2-arachidonoyl glycerol hydrolase activity( Reference Blankman, Simon and Cravatt 6 ). Within the brain, ABHD6 seems to be expressed by neurones and often to be located postsynaptically, close to neurones that express presynaptic CB1 receptors, whereas ABHD12 seems to be expressed by non-neuronal cells, for example microglia( Reference Savinainen, Saario and Laitinen 7 ).
Interestingly, there is evidence that some of the metabolites formed from endogenously produced anandamide or 2-arachidonoyl glycerol by enzymes other than FAAH and MAG lipase are pharmacologically active. For example, one COX-2 metabolite of anandamide is PGH2 ethanolamide, which is further metabolised to J-series PG that have been found to induce apoptosis in COX-2 overexpressing tumourigenic keratinocytes, an effect that could be enhanced by an irreversible FAAH inhibitor (URB597;( Reference Kuc, Jenkins and Van Dross 8 ) (Fig. 1a). It has also been found that COX-2 metabolises 2-arachidonoyl glycerol to the hyperalgesic prostanoid, PGE2 glycerol ester, an observation that has prompted the hypothesis that COX-2 could act as an enzymatic switch with the capacity to convert 2-arachidonoyl glycerol from an antinociceptive mediator to a pronociceptive prostanoid( Reference Hu, Bradshaw and Chen 9 ).

Fig. 1. Structures of a selection of inhibitors of (a) fatty acid amide hydrolase (FAAH), (b) monoacylglycerol lipase (MAG lipase), (c) dual inhibitors of FAAH and MAG lipase and (d) the cellular uptake of anandamide.
FAAH inhibitors other than URB597 (Org231295) include both the reversible inhibitor, OL135 (Fig. 1a) and O-1887, which, like URB597, is an irreversible inhibitor( Reference Di Marzo 10 – Reference Pertwee 12 ). These can all inhibit FAAH in vitro at concentrations that lie in the low nanomolar range and do not displace radiolabelled ligands from CB1 receptors or (where this has been investigated) from CB2 receptors. It is noteworthy, however, that the pharmacological profiles of most FAAH inhibitors have not been completely investigated. Just a few additional examples of the many FAAH inhibitors that have now been developed include URB532 (Fig. 1a), AM374 (palmitylsulphonyl fluoride), N-arachidonoyl glycine and N-arachidonoyl serotonin( Reference Pertwee 2 ), JNJ1661010 and CAY10401( Reference Petrosino and Di Marzo 13 ), AM3506 and AM5206( Reference Godlewski, Alapafuja and Bátkai 14 , Reference Naidoo, Nikas and Karanian 15 ), ST4070( Reference Gattinoni, De Simone and Dallavalle 16 , Reference Caprioli, Coccurello and Rapino 17 ), PF3845( Reference Ahn, Johnson and Mileni 18 ) and PF04457845, a particularly FAAH-selective structural analogue of PF3845( Reference Johnson, Stiff and Lazerwith 19 ) (Fig. 1a).
Evidence has already emerged from animal experiments that FAAH inhibitors are effective at ameliorating signs of acute, inflammatory, visceral and neuropathic pain( Reference Pertwee 2 , Reference Alvarez-Jaimes and Palmer 4 , Reference Petrosino and Di Marzo 13 , Reference Caprioli, Coccurello and Rapino 17 – Reference Okine, Norris and Woodhams 24 ), and that URB597 can ameliorate the signs of both osteoarthritic pain( Reference Schuelert, Johnson and Oskins 25 ), and referred hyperalgesia induced by bladder inflammation( Reference Merriam, Wang and Hillard 26 ). Animal experiments have also yielded data suggesting that FAAH inhibitors might have a number of other therapeutic applications (Table 1). Importantly, there is good evidence that anandamide displays significant potency not only as a cannabinoid CB1 and CB2 receptor agonist, but also at activating or inhibiting certain voltage-gated ion channels, at enhancing the activation of NMDA and glycine ligand-gated ion channels, at blocking transient receptor potential (TRP) M8 cation channels and at allosterically antagonising the activation of 5-hydroxytryptamine3 and nicotinic acetylcholine receptors( Reference Pertwee, Howlett and Abood 1 ). It is also well-established, first, that anandamide can activate TRPV1 and TRPA1 cation channels, albeit with less potency than it activates cannabinoid receptors( Reference Pertwee, Howlett and Abood 1 ), and second, that FAAH can metabolise pharmacologically active endogenous compounds other than anandamide, just one example being oleoylethanolamide which is not a cannabinoid receptor ligand but can activate both TRPV1 cation channels and the PPARα ( Reference Piscitelli and Di Marzo 49 ). It is possible too that reductions in signs of nicotine-induced reward evoked by URB597 may be PPARα-mediated since this FAAH inhibitor seems to produce this effect mainly by elevating oleoylethanolamide and palmitoylethanolamide, which is also an endogenous PPARα ligand( Reference Scherma, Panlilio and Fadda 40 ). It is noteworthy, therefore, that there have been several investigations in which a selective CB1 or CB2 receptor antagonist( Reference Caprioli, Coccurello and Rapino 17 , Reference Miller, Picker and Umberger 23 , Reference Okine, Norris and Woodhams 24 , Reference Khasabova, Khasabov and Paz 50 , Reference Starowicz, Makuch and Osikowicz 51 ), or genetic deletion of the CB1 receptor( Reference Booker, Kinsey and Abdullah 52 ), has been found to attenuate signs of antinociception induced by URB597, PF3845 or ST4070 in rodent models of visceral, inflammatory or neuropathic pain. In two of these investigations, however, the TRPV1 antagonist, 5′-iodoresiniferatoxin,( Reference Starowicz, Makuch and Osikowicz 51 ), or the PPARα antagonist, GW6471( Reference Caprioli, Coccurello and Rapino 17 , Reference Okine, Norris and Woodhams 24 ), was also found to block at least some antinociceptive effects of URB597 or ST4070, whereas in another of these investigations no such antagonism was detected( Reference Booker, Kinsey and Abdullah 52 ). In addition, there have been reports that a CB1 receptor antagonist can block AM3506-induced inhibition of abnormal gastrointestinal motility and faecal output in mice( Reference Bashashati, Storr and Nikas 31 ), and also URB597-induced reductions of vomiting in shrews and of signs of nausea in rats( Reference Parker, Rock and Limebeer 45 ), and that both a CB1 and a CB2 receptor antagonist can block URB597-induced inhibition of scratching behaviour that had been evoked by 5-hydroxytryptamine in rats( Reference Spradley, Davoodi and Gee 36 ).
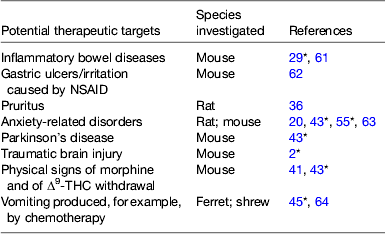
Table 1. Some potential therapeutic targets other than pain identified in preclinical investigations for fatty acid amide hydrolase inhibitors

IBS, irritable bowel syndrome; l-DOPA, l−3,4-dihydroxyphenylalanine; NSAID, non-steroidal anti-inflammatory drug; Δ9-THC, Δ9-tetrahydrocannabinol.
* Review article.
Moving on to MAG lipase inhibitors, these include (Fig. 1b) the non-competitive/irreversible inhibitors, URB602 and JZL184, and the reversible inhibitor, OMDM169( Reference Petrosino and Di Marzo 13 ). Like inhibitors of FAAH, MAG lipase inhibitors display antinociceptive activity in a number of different animal models that for one or more of these compounds include models of acute, visceral, inflammatory, neuropathic and/or bone cancer pain( Reference Alvarez-Jaimes and Palmer 4 , Reference Petrosino and Di Marzo 13 , Reference Busquets-Garcia, Puighermanal and Pastor 20 – Reference Kinsey, Long and O'Neal 22 , Reference Mulvihill and Nomura 43 , Reference Bisogno, Ortar and Petrosino 53 – Reference Long, Li and Booker 57 ). There is evidence too, that in rats, MAG lipase inhibitors can reduce signs of inflammatory pain not only when administered systemically but also when injected directly into the spinal cord or inflamed hind paws( Reference Guindon, Desroches and Beaulieu 58 – Reference Guindon, Guijarro and Piomelli 60 ). Moreover, results obtained from animal experiments with JZL184 suggest that, like FAAH inhibitors, MAG lipase inhibitors have several other potential therapeutic applications (Table 2). There is evidence too that MAG lipase inhibitors might be effective against breast, ovarian, skin and prostate cancer( Reference Mulvihill and Nomura 43 ). Thus, for example, it has been found that JZL184 can reduce prostate cancer (PC3) cell migration, invasion and survival in vitro and slow PC3 tumour xenograft growth in immune-deficient SCID mice, although interestingly, not in nude mice, probably because its inhibitory effect on MAG lipase was less in the tumours grown in nude mice than in those grown in SCID mice( Reference Nomura, Lombardi and Chang 65 ). Evidence has also emerged that JZL184 and URB602 can protect neurons from β-amyloid peptide-induced neurodegeneration and apoptosis, raising the possibility that Alzheimer's disease could be another suitable therapeutic target for the MAG lipase inhibitors( Reference Mulvihill and Nomura 43 , Reference Chen, Zhang and Chen 66 – Reference Piro, Benjamin and Duerr 68 ).
Table 2. Some potential therapeutic targets other than pain identified in preclinical investigations for monoacylglycerol lipase inhibitors
NSAID, non-steroidal anti-inflammatory drugs; Δ9-THC, Δ9-tetrahydrocannabinol.
* Review article.
Like anandamide, 2-arachidonoyl glycerol, does not target only cannabinoid CB1 and CB2 receptors. Thus, there is evidence first, that at concentrations similar to those at which it activates these receptors, 2-arachidonoyl glycerol can inhibit KV4.3 voltage-gated ion channels and allosterically antagonise the activation of nicotinic acetylcholine receptors, and second, that at higher concentrations, it can also target a number of other receptors or ion channels, including glycine, γ-aminobutyric acidA and adenosine A3 receptors and certain voltage-gated calcium channels( Reference Pertwee, Howlett and Abood 1 , Reference Sigel, Baur and Rácz 69 ). Even so, at least some of the effects that the MAG lipase inhibitors have been reported to produce in vivo, appear to have been partly or wholly cannabinoid-receptor mediated. More specifically, there have been reports that the CB1-selective antagonist, AM251 reduces the ability of JZL184 to attenuate vomiting in shrews and signs of nausea in rats( Reference Sticht, Long and Rock 64 ), that AM251 and the CB2-selective antagonist, AM630, both prevent JZL184-induced reductions of 5-hydroxytryptamine-evoked scratch bouts in rats( Reference Spradley, Davoodi and Gee 36 ), and that these two antagonists can also prevent JZL184 from reducing signs of colitis in mice( Reference Alhouayek, Lambert and Delzenne 61 ). Evidence has also been obtained from experiments performed with CB1 and CB2 receptor-deficient mice, that reductions in signs of neuropathic pain induced by JZL184 were mediated by CB1 but not CB2 receptors( Reference Kinsey, Long and Cravatt 21 ). In contrast, the production of this effect by the FAAH inhibitor, PF3845, seemed to depend on the activation of both the CB1 and CB2 receptors. It is noteworthy too, that Busquets-Garcia et al. ( Reference Busquets-Garcia, Puighermanal and Pastor 20 ) have found that an anxiolytic-like effect produced by JZL184 in mice seemed to be CB2-receptor mediated since it could be abolished firstly, by the CB2-selective antagonists, SR144528 and AM630, but not by the CB1-selective antagonist, SR141716A (rimonabant), and secondly, by genetic deletion of the CB2 receptor but not of the CB1 receptor. On the other hand, the FAAH inhibitor, URB597, produced an anxiolytic-like effect in mice that appeared to be CB1 but not CB2 receptor-mediated.
The evidence that some of the apparently beneficial effects of FAAH and MAG lipase inhibitors appear to be partly or wholly cannabinoid CB1 receptor-mediated, prompts the question of whether these inhibitors also produce some or all of the unwanted effects that can result from direct CB1 receptor activation. It is noteworthy, therefore, that there have been reports that unlike direct CB1 agonists, it is possible for FAAH inhibitors to produce antinociception in mice at doses that do not also induce hypomotility, hypothermia, catalepsy or hyperphagia( Reference Cravatt and Lichtman 70 – Reference Lichtman, Leung and Shelton 72 ). It is also unlikely that a FAAH inhibitor would share the ability of a direct CB1 receptor agonist to induce physical or psychological dependence. Thus, repeated administration of one FAAH inhibitor, PF3845, has been found not to produce signs of physical dependence in mice( Reference Schlosburg, Blankman and Long 73 ), and the results obtained with another FAAH inhibitor, URB597, in experiments with squirrel monkeys, suggest that such inhibitors do not possess reinforcing properties and would not trigger relapse to drug abuse( Reference Justinova, Mangieri and Bortolato 74 ). Since tolerance can develop to antinociceptive effects of direct cannabinoid receptor agonists( Reference Schlosburg, Blankman and Long 73 ), it is noteworthy too, however, that there has been a report that tolerance appeared to develop to the ability of URB597 to attenuate inflammatory pain behaviour in rats, although not to its ability to inhibit FAAH, when this compound was administered repeatedly( Reference Okine, Norris and Woodhams 24 ).
With regard to MAG lipase inhibitors, there has been a report that JZL184 shares the ability of direct CB1 receptor agonists to induce both hypothermia and hypomotility, although not catalepsy, at an antinociceptive dose( Reference Long, Li and Booker 57 ). Moreover, when it was administered repeatedly to mice, this MAG lipase inhibitor was found to produce physical dependence, as indicated by SR141716A-precipitated paw fluttering, and to cause tolerance to develop both to its own antinociceptive effect, unless it was administered at a low antinociceptive dose, and to that of the direct CB1/CB2 receptor agonists, Δ9-tetrahydrocannabinol and R-(+)-WIN55212( Reference Ghosh, Wise and Chen 56 , Reference Schlosburg, Blankman and Long 73 ). In line with some of these observations is the finding that both JZL184 and the direct CB1/CB2 receptor partial agonist, Δ9-tetrahydrocannabinol, could reduce four naloxone precipitated physical withdrawal signs in morphine-dependent mice( Reference Ramesh, Ross and Schlosburg 41 ). In contrast, only two of these withdrawal signs could be attenuated by the FAAH inhibitor, PF3845. Finally, the data obtained by Busquets-Garcia et al. ( Reference Busquets-Garcia, Puighermanal and Pastor 20 ) from mouse experiments suggest that a dose of JZL184 that induced apparent CB2 receptor-mediated reductions in anxiety, did not affect memory, whereas memory was impaired by URB597 at a dose that induced apparent CB1 receptor-mediated anxiolysis. No tolerance developed to the anxiolytic-like effect of JZL184 or URB597, or to the memory-impairing effect of URB597, when either of these two compounds was administered repeatedly.
Clearly, further research is required to establish whether a FAAH-inhibitor is likely to have a significantly higher or lower benefit-to-risk ratio than a MAG lipase inhibitor for each of the potential therapeutic applications that they have in common. It is noteworthy, however, that there is already strong evidence that in order to optimise the benefit-to-risk ratio of any new medicine that acts by inhibiting endocannabinoid metabolism, it will be important to ensure that this medicine does not inhibit both these enzymes at the same time since it is likely that the therapeutically beneficial and adverse effects produced by dual inhibitors of FAAH and MAG lipase would closely resemble those that are produced by direct cannabinoid receptor agonists. Thus, signs of central CB1 receptor activation that were very similar to those produced by a direct CB1 receptor agonist and included hypomotility, antinociception and catalepsy, have been found to be displayed by mice in response to such dual inhibition, by certain organophosphorus nerve agents( Reference Nomura, Blankman and Simon 75 ), and by JZL195 (Fig. 1c), a compound that inactivates both FAAH and MAG lipase in vivo with high efficacy, and is also an inhibitor of ABHD6( Reference Alvarez-Jaimes and Palmer 4 , Reference Long, Nomura and Vann 76 ). More importantly, these dual inhibitor-induced effects were blocked by pretreatment with a CB1-selective antagonist: either AM251 or SR141716A. It has also been found that mice treated with JZL195 displayed signs of impaired short-term spatial memory that were similar in magnitude to those produced by the direct CB1/CB2 receptor agonist, Δ9-tetrahydrocannabinol, and could be prevented by SR141716A( Reference Wise, Long and Abdullah 77 ). A similar effect on memory was induced by the MAG lipase inhibitor, JZL184, in mice from which FAAH had been genetically deleted( Reference Wise, Long and Abdullah 77 ). Interestingly, however, another dual inhibitor of FAAH and MAG lipase, AM6701 (Fig. 1c), has been reported to reduce both severity of seizures and impairment of rotarod and balance beam performance induced in rats by kainic acid with significantly greater efficacy than AM6702, a compound that is significantly more potent at inhibiting FAAH than MAG lipase( Reference Naidoo, Karanian and Vadivel 78 ).
A dual inhibitor of FAAH and MAG lipase might of course have an acceptable benefit-to-risk ratio if, for example, its distribution is restricted to one or more particular regions of the body. Indeed, there is already evidence that one possible strategy would be to administer a dual inhibitor to the skin for the treatment of cancer pain( Reference Khasabova, Chandiramani and Harding-Rose 79 ). An alternative possibility would be to administer a peripherally-restricted dual inhibitor of FAAH and MAG lipase, or indeed to co-administer a FAAH inhibitor and a MAG lipase inhibitor, either or both of which are peripherally restricted. One example of a peripherally-restricted FAAH inhibitor that has already been developed is URB937( Reference Moreno-Sanz, Barrera and Guijarro 80 ), a compound that has been reported to exert profound antinociceptive effects in rodent models of visceral, inflammatory and neuropathic pain( Reference Clapper, Moreno-Sanz and Russo 81 ). Since the hydrolases, MAG lipase, ABHD6 and ABHD12, most probably metabolise distinct pools of 2-arachidonoyl glycerol in vivo ( Reference Blankman, Simon and Cravatt 6 ), further research that explores the potential therapeutic advantages of selectively inhibiting just one or other of these enzymes, or indeed of simultaneously inhibiting two or even all three of these enzymes, is also clearly warranted. It is noteworthy, therefore, that a selective inhibitor of ABHD6 was developed in the same year that this enzyme was found to metabolise 2-arachidonoyl glycerol( Reference Alvarez-Jaimes and Palmer 4 , Reference Blankman, Simon and Cravatt 6 , Reference Li, Blankman and Cravatt 82 ).
Evidence has also emerged from animal experiments not only that administration of a FAAH inhibitor in combination with a COX inhibitor/non-steroidal anti-inflammatory drug (NSAID) would be effective for the amelioration of inflammatory and neuropathic pain( Reference Sasso, Bertorelli and Bandiera 34 ) and visceral pain( Reference Fowler 32 , Reference Naidu, Booker and Cravatt 33 ), but also, as already mentioned, that FAAH inhibition lowers the unwanted ulcerogenic activity of COX inhibitors/NSAID( Reference Fowler 32 – Reference Sasso, Bertorelli and Bandiera 34 , Reference Favia, Habrant and Scarpelli 83 ). It is noteworthy, therefore, that carprofen, a compound that has been reported to produce fewer ulcerogenic side effects in human subjects than other NSAID, has been found to be a multi-inhibitor of COX-1, COX-2 and FAAH( Reference Favia, Habrant and Scarpelli 83 ). It is noteworthy, too, first that the peripherally restricted FAAH inhibitor, URB937, has been found in mice both to interact synergistically with the NSAID, indomethacin, to reduce signs of inflammatory and neuropathic pain and to lower the number and severity of indomethacin-induced gastric lesions( Reference Sasso, Bertorelli and Bandiera 34 ), and second, that the MAG lipase inhibitor, JZL184, has also been found to protect against NSAID-induced gastric damage in mice( Reference Kinsey, Nomura and O'Neal 62 ). Other potential adjunctive therapeutic strategies include the combined administration of a FAAH inhibitor, with a COX-2 inhibitor and a low dose of a glucocorticoid for rheumatoid arthritis( Reference Lowin, Zhu and Dettmer-Wilde 84 ), with palmitoylethanoamide for the treatment of melanoma( Reference Hamtiaux, Masquelier and Muccioli 85 ), with morphine for relieving visceral pain( Reference Miller, Picker and Umberger 23 ), and with an inhibitor of the metabolism of endogenous opioids (enkephalins) for the alleviation of inflammatory and neuropathic pain( Reference Roques, Fournié-Zaluski and Wurm 86 ).
Finally, there has been one recent published report about a clinical trial performed with the FAAH inhibitor, PF04457845( Reference Huggins, Smart and Langman 87 ). This was a multicentre, randomised, double-blind, placebo-controlled trial in which patients experiencing pain due to osteoarthritis of the knee received repeated doses of PF04457845, which has a good human safety profile( Reference Huggins, Smart and Langman 87 , Reference Li, Winter and Arends 88 ), of naproxen which served as a positive control, or of placebo. Unexpectedly, it was found that although PF04457845 did seem to inhibit FAAH, as indicated by its ability to elevate the plasma levels of anandamide, palmitoylethanolamide, oleoylethanolamide and linoleoylethanolamide in patients, it did not share the ability of naproxen to reduce osteoarthritic pain in this trial. Given the many positive results that have been obtained with FAAH inhibitors in animal models of pain, including rat and guinea-pig models of osteoarthritic pain( Reference Schuelert, Johnson and Oskins 25 ), the inability of PF04457845 to relieve human osteoarthritic pain at a dose that does seem to inhibit FAAH prompts an urgent need for further research.
Potential therapeutic applications for inhibitors of endocannabinoid cellular uptake
A number of compounds that can inhibit the cellular uptake of anandamide have been developed. These include AM404, VDM11 and UCM707 (Fig. 1d), as well as OMDM1, OMDM2 and LY2183240( Reference Pertwee 2 , Reference Di Marzo 10 ), one or more of which has been found to decrease cancer cell proliferation and tumour growth and to be effective in animal models against signs of pain, multiple sclerosis, excitotoxicity, Parkinson's disease, Huntington's disease, anxiety, cholera toxin-induced diarrhoea and colitis( Reference Pertwee 2 , Reference Di Marzo 10 , Reference Miller, Picker and Umberger 23 ). In addition, evidence has recently emerged that reinstatement of nicotine-seeking behaviour can be inhibited by both VDM11 and AM404 in rats, and that the latter compound can prevent the development of nicotine dependence in rats as well( Reference Gamaleddin, Guranda and Goldberg 89 , Reference Scherma, Justinová and Zanettini 90 ). Evidence has also been obtained, from mouse experiments, that AM404 might be effective against obsessive compulsive disorder( Reference Gomes, Casarotto and Resstel 37 ). In addition to inhibiting the cellular uptake of anandamide, AM404 can activate and desensitise TRPV1 receptors, and inhibit COX-1, COX-2 and PG synthesis( Reference Piscitelli and Di Marzo 49 , Reference Reyes-Cabello, Alen and Gómez 91 ). Even so, this compound has been found to reduce signs of visceral pain in mice in a manner that seems to be CB1 receptor-mediated( Reference Miller, Picker and Umberger 23 ). Importantly, LY2183240 is an inhibitor not only of endocannabinoid cellular uptake but also of FAAH and MAG lipase, and VDM11, UCM707 and AM404, which is a FAAH substrate, also have the ability to reduce the metabolism of anandamide and other acylethanolamides by FAAH( Reference Di Marzo 10 , Reference Scherma, Justinová and Zanettini 90 ).
Although endocannabinoids clearly do pass through cell membranes, the question of whether or not this process is mediated by a transporter has been a topic of debate for many years( Reference Piscitelli and Di Marzo 49 ). It is noteworthy, therefore, that there is now evidence for the existence of an anandamide-selective intracellular binding protein that may facilitate first, the cellular uptake of anandamide by reducing the free concentration of anandamide within cells; second, the translocation of anandamide to intracellular organelles where FAAH-1 is located, and third, the release of anandamide from cells by increasing its intracellular transfer from its sites of biosynthesis within cell membranes( Reference Piscitelli and Di Marzo 49 , Reference Fu, Bottegoni and Sasso 92 , Reference Marsicano and Chaouloff 93 ). This binding protein has been named FAAH-like anandamide transporter. ARN272, a compound that inhibits both the binding of anandamide to FAAH-like anandamide transporter in a competitive manner, and the intracellular accumulation of this endocannabinoid, has been found to attenuate signs of inflammatory pain in mice( Reference Fu, Bottegoni and Sasso 92 ). There has been a report too that SB-Fl-26, an inhibitor of other intracellular transporters of anandamide that are known as fatty acid binding proteins, also displays antinociceptive activity in a mouse model of inflammatory pain( Reference Berger, Ralph and Kaczocha 94 ).
Conclusions and future directions
The present paper has reviewed evidence published up to March 2013, that signs and symptoms of a number of disorders could be ameliorated by administering an inhibitor of the anandamide-metabolising enzyme, FAAH, of the 2-arachidonoyl glycerol-metabolising enyzyme, MAG lipase or of endocannabinoid cellular uptake. Although the published findings obtained with these inhibitors are very promising, they have come almost entirely from experiments with animals, prompting an urgent need for clinical trials that build on these findings by seeking out disorders in which significant beneficial effects could be achieved by (i) administering selective inhibitors of FAAH, MAG lipase or endocannabinoid cellular uptake; (ii) simultaneously inhibiting FAAH and MAG lipase, for example with a dual inhibitor; (iii) targeting particular regions of the body, for example, by direct application of one or more of these inhibitors to the skin, or by administering peripherally restricted inhibitors; and (iv) inhibiting FAAH or MAG lipase simultaneously with either or both COX-1 and COX-2. The need for such research with patients is strengthened by the findings already obtained in clinical trials, first, that the FAAH inhibitor, PF04457845, did not reduce osteoarthritic pain, and second, that concomitant inhibition of COX-1, COX-2 and FAAH produces fewer ulcerogenic side effects in human subjects than inhibition of just COX-1 and COX-2 by an NSAID. In addition, it will be important to search for potential therapeutic strategies that exploit: (i) the existence of at least two subtypes of FAAH; FAAH-1 and FAAH-2, and (ii) the recently discovered presence in the brain of a new set of endocannabinoids: lipoxin A4, which behaves as an allosteric enhancer of anandamide-induced activation of the CB1 receptor( Reference Pamplona, Ferreira and Menezes-de-Lima 95 , Reference Pertwee 96 ), and an endogenous peptide, named Pepcan-12, that can weaken CB1 receptor activation by direct agonists, seemingly by acting as a negative allosteric modulator at these receptors( Reference Bauer, Chicca and Tamborrini 97 ).
Acknowledgements
R. G. P. is grateful for financial support from both GW Pharmaceuticals and the National Institute on Drug Abuse.
Financial Support
The work was supported by Research grants from the National Institute on Drug Abuse (grant numbers DA-009789 and DA-03672) and funding from GW Pharmaceuticals.
Conflicts of Interest
None.



