



Introduction
We currently lack tools for effective genome-wide characterization of schistosomes from human populations for two main reasons. First, there is the practical problem of obtaining suitable material for genomic analyses: schistosome adults live in the blood vessels and only eggs expelled in feces (Schistosoma mansoni and japonicum) or urine (Schistosoma haematobium) are available from patients. Microscopic miracidium larvae (70 × 140 µ m) hatched from these eggs can be used to infect snails and the cercaria larvae produced can be used to infect laboratory rodents to recover adult worms, but this approach is cumbersome, ethically questionable and imposes strong selection and potential bias (Gower et al., Reference Gower2007). An alternative approach is to hatch these larvae and preserve them individually on FTA cards for genetic analysis (Gower et al., Reference Gower2007). This approach has been widely used and has greatly improved our understanding of many aspects of schistosome population biology (Steinauer et al., Reference Steinauer, Blouin and Criscione2010; Webster et al., Reference Webster2012; Gower et al., Reference Gower2013, Reference Gower2017). However, these studies can be severely constrained by the minute amount of DNA and therefore the relatively small number of genetic markers that can be successfully multiplexed, PCR amplified and analysed from a single miracidium. Typically these studies utilize just 8–20 microsatellite markers (Gower et al., Reference Gower2011, Reference Gower2013, Reference Gower2017; Glenn et al., Reference Glenn2013; Steinauer et al., Reference Steinauer2013; Aemero et al., Reference Aemero2015; Webster et al., Reference Webster2015) or 1–3 gene regions (Van den Broeck et al., Reference Van den Broeck2015; Li et al., Reference Li2017). The use of FTA cards has allowed the creation of large archived collections of FTA-preserved miracidia maintained at the Schistosomiasis Collection at the Natural History Museum (SCAN) (Emery et al., Reference Emery2012). SCAN currently houses around 300 000 miracidia collected from over 7000 people from 14 countries, sampled within the past decade, providing valuable geographical and temporal populations of samples for molecular analysis.
The size and complex architecture of schistosome genomes provide an additional obstacle. Whole genome sequences of the three principal schistosome species infecting humans (S. mansoni, S. haematobium and S. japonicum) are now available, but these are large (363–400 Mb), and riddled with transposable elements and repeats, which comprise 40–50% of the genome and make alignment problematic (Berriman et al., Reference Berriman2009; Zhou et al., Reference Zhou2009; Protasio et al., Reference Protasio2012; Young et al., Reference Young2012). The large genome size also makes whole genome sequencing from populations prohibitively expensive, even with falling sequencing costs. Furthermore, the coding regions of the genome (the exome) that are of primary interest for many analyses measure ~15 Mb and constitute just 4% of the total genome (Young et al., Reference Young2012).
Exome capture methods utilize RNA baits that hybridize with the targeted exome sequences, so they can be isolated from non-coding DNA. This approach has been widely used to rapidly and economically sequence thousands of human exomes and identify coding variants associated with disease (Rabbani et al., Reference Rabbani, Tekin and Mahdieh2014; Pehlivan et al., Reference Pehlivan2014; Shaw et al., Reference Shaw2017). There are several attractive features of exome sequencing for schistosome miracidia: (i) this approach is scalable to working with hundreds of individual miracidia, (ii) repetitive regions of the genome that are problematic to align are removed, (iii) contaminating sequences amplified from FTA cards (e.g. bacteria from fecal matter and water) are eliminated, and (iv) reducing the genome size from 363 to 15 Mb allows sequencing to high read depth, providing robust scoring of variable sites. We have previously described an exome capture approach that we used for whole genome amplified DNA prepared from adult S. mansoni (Chevalier et al., Reference Chevalier2014). Here we adapt this approach for single miracidia. We describe methods for (i) whole genome amplification (WGA) of miracidia DNA directly from FTA cards, (ii) qPCR methods for quantifying of schistosome DNA amplified and (iii) exome capture, sample multiplexing and sequencing of amplified DNA. We apply these methods to a subset of miracidia from SCAN to demonstrate the utility of these methods. Furthermore, we describe protocols for both S. mansoni and S. haematobium, the two species responsible for more than 99% of human infections (Hotez et al., Reference Hotez, Jamison, Breman, Measham, Alleyne, Claeson, Evans, Jha, Mills and Musgrove2006).
Materials and methods
Ethics statement
EU-CONTRAST specimens were collected in line with project ethical approval granted by ethical committees of the Ministry of Health Dakar, Senegal, the Niger National Ethical Committee, Comité National d'Ethique, Cameroon. For specimens from the Schistosomiasis Consortium for Operational Research and Evaluation (SCORE), ethical approvals were granted by Royal Veterinary College 2015 1327; Imperial College Research Ethics Committee and SCI Ethical approval (EC no: 03.36. R&D no: 03/SB/033E); the National Institute for Medical Research (NIMR, reference no. NIMR/HQ/R.8a/Vol. IX/1022); Zanzibar Medical Research Ethics Committee in Stonetown, Zanzibar (ZAMREC, reference no. ZAMREC 0003/Sept/011); University of Georgia Institutional Review Boards, Athens, GA (2011-10353-1); Niger Republic National Consulate (reference no. 012/2010/CCNE).
All local requirements were met, including obtaining written informed consent from adults (including parents/legal guardians of children included in the studies) and followed by obtaining the assent from children included. Following sampling, a praziquantel treatment (40 mg.kg−1) was offered to infected participants.
Field collection sites
Schistosoma mansoni miracidia were collected as part of EU-CONTRAST activities from individual patients in three different West African countries in 2007: 27 patients from two locations in Senegal, 17 patients from two locations in Niger and six patients from one location in Cameroon (Supplementary File 1) (Gower et al., Reference Gower2011; Webster et al., Reference Webster2013). Additionally, S. mansoni miracidia were collected as part of the SCORE programme from 64 children in seven villages on the shores of Lake Victoria in Tanzania (East Africa) in January 2012 (Ezeamama et al., Reference Ezeamama2016) (Supplementary File 1).
Schistosoma haematobium miracidia were collected through the SCORE Zanzibar Elimination of Schistosomiasis Transmission (ZEST) activities in 2011 from 80 children in 26 locations of the Zanzibar archipelago (East Africa) and in 2013 from 57 patients in 10 locations of Niger (West Africa) (Supplementary File 1).
All samples were transferred to SCAN for checking, curation and storage either immediately (SCORE), or at the close of the project (EU-CONTRAST).
Collection of miracidia
Our methods for recovery of S. mansoni eggs followed Visser and Pitchford (Reference Visser and Pitchford1972) and Gower et al. (Reference Gower2013) with some minor differences. In brief, (i) each S. mansoni infected stool sample (approximately 2 g) was homogenized with a plastic spatula through a 212 µ m wire mesh sieve (Endecotts Ltd, London, UK) and washed through with approximately 1 L of locally available bottled water, (ii) the tray contents were transferred to the inner nylon mesh (200 µ m pore size) of a Pitchford funnel assembly (Visser and Pitchford, Reference Visser and Pitchford1972), washed with additional water (up to an additional 1 L), and the inner mesh removed from the assembly, (iii) the washed, filtered homogenate was drained into a 100 × 15 mm Petri dish (BD Biosciences, Erembodegem, Belgium) by opening the tap at the base of the Pitchford funnel. The Petri dish containing the homogenate was then left in bright ambient light (not direct sunlight) to allow hatching of miracidia.
Schistosoma haematobium eggs were concentrated from each infected urine by sedimentation, then rinsed in bottled mineral water before transfer into a clean Petri dish containing mineral water and exposed to light to facilitate hatching (Webster et al., Reference Webster2012).
Both miracidia from S. mansoni and S. haematobium were captured individually, under a binocular microscope, in 3 µL of water and spotted onto an indicating Whatman FTA Classic Indicating card (GE Healthcare Life Sciences, Amersham, UK) using a 20 µL micropipette. Spotted samples (up to approximately 80 per card) can be easily located on the cards because the pink dye turns white after water contact. The cards were then allowed to dry for 1 h at room temperature before being stored in a sealed plastic bag and then shipped to UK, ultimately to be stored in the SCAN repository.
Preparation of FTA samples for WGA
For each sample prepared, a 2 mm diameter disc was removed from the centre of the dye-cleared area using a 2 mm Harris Micro-punch (GE Healthcare Life Sciences, Amersham, UK). This 2 mm disc corresponds to the entire spot containing the whole miracidium. Each punch was placed individually in 500 µL Matrix screw cap two-dimensional barcode storage tubes (Thermo Fisher Scientific, Hemel Hempstead, UK) and shipped to Texas Biomedical Research Institute for further preparation.
The punches were individually transferred into 1.5 mL sterile microtubes using sterile tips. Punches were washed three times with FTA Purification Reagent (GE Healthcare Life Sciences, Logan, Utah, USA) then rinsed two times with TE−1 buffer (10 mm Tris, 0.1 mm EDTA, pH 8). Washing and rinsing steps were performed by adding 200 µL of solution in each tube followed by 5 min of incubation on a nutating mixer (24 RPM) at room temperature and then discarding the solution while minimizing contact between the pipette tip and the punch. Punches were finally dried in tubes during 10 min at 56 °C in a dry bath incubator (Chevalier et al., Reference Chevalier2016).
Whole genome amplification
We conducted WGA on 1–4 miracidia from each patient from all the sites sampled until a positive WGA was obtained. We performed WGA on each punch using the Illustra GenomiPhi V2 DNA Amplification kit (GE Healthcare Life Sciences, Logan, Utah, USA). Punches were transferred into 0.2 mL sterile tubes using a sterile tip. Reactions were performed following the manufacturer's instructions, immersing each punch in 9 µL of sample buffer and keeping tubes on ice after the denaturation step (3 min at 95 °C) while adding a mix of 9 µL of reaction buffer and 1 µL of Phi29 polymerase. After the amplification step (2 h 30 min at 30 °C and 10 min at 65 °C), we added 130 µL of sterile water into the reaction tube and purified the 150 µL of amplified genome with the SigmaSpin™ Sequencing reaction Clean-up (Sigma-Aldrich, Laramie, Wyoming, USA), following the manufacturer protocol. We quantified purified samples using the Qubit dsDNA BR assay (Invitrogen, Grand Island, New York, USA).
Real-time quantitative PCR on schistosome WGA samples
We performed real-time quantitative PCR (qPCR) reactions to estimate the proportion of schistosome genome in each WGA sample. This was done because FTA samples can contain contaminant DNA, such as human or bacterial DNA, which is co-amplified with schistosome DNA during the WGA step. In this assay, we amplified the S. mansoni α-tubulin 1 (sat-1) gene, which is present in low copy (gene number: Smp_090120; accession number: M80214) (Webster et al., Reference Webster1992) or the putative S. haematobium α-tubulin 2. The latter was identified by performing a blastn (v2.2.29) using the S. mansoni α-tubulin (Duvaux-Miret et al., Reference Duvaux-Miret1991; accession number: S79195) against the S. haematobium reference genome (SchHae_1.0; assembly accession number: GCA_000699445.1). Reactions were performed in duplicate using the AB1 prism 7900HT Sequence Detection system (Applied Biosystems, Carlsbad, California, USA) as follows: 95 °C for 10 min, then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Duplicate reactions showing a difference in C T greater than one were rerun. We examined the melting curve (60–95 °C) at the end of each assay to verify the uniqueness of the PCR products generated. The reaction mixture consisted of 5 µL of SYBR Green MasterMix (Applied Biosystems, Carlsbad, California, USA), 0.3 µL of 10 µM forward primer (S. mansoni: CGAAATTGGAGTTTGCTGTGT; S. haematobium: GGTGGTACTGGTTCTGGTTT) and 10 µM reverse primer (S. mansoni: TGTAGGTTGGACGCTCTATATC; S. haematobium: AAAGCACAATCCGAATGTTCTAA) amplifying 229 bp of the sat-1 gene for S. mansoni and 178 bp of the α-tubulin 2 for S. haematobium, 3.4 µL of sterile water and 1 µL of total DNA template (normalized at 20 ng.µL−1). We plotted standard curves using seven dilutions of a purified sat-1 PCR product for S. mansoni (sat-1 copies.µL−1: 2.19 × 101, 2.19 × 102, 2.19 × 103, 2.19 × 104, 2.19 × 105, 2.19 × 106, 2.19 × 107) or seven dilutions of a purified α-tubulin 2 PCR product for S. haematobium (α-tubulin 2 copies.µL−1: 1.29 × 101, 1.29 × 102, 1.29 × 103, 1.29 × 104, 1.29 × 105, 1.29 × 106, 1.29 × 107). The number of sat-1 or α-tubulin 2 copies in each sample was estimated according to the standard curve.
Exome library preparation and sequencing
We captured schistosome (S. mansoni or S. haematobium) exomes using the SureSelectXT2 Target Enrichment System (Agilent) according to the manufacturer's protocol. For each library (i.e. each sample), we sheared 1 µg of WGA DNA by adaptive focused acoustics (Duty factor: 10%; Peak Incident Power: 175; Cycles per Burst: 200; Duration: 180 s) in AFA tubes, using a Covaris S220 instrument with SonoLab software version 7 (Covaris, Inc., Woburn, Massachussetts, USA), to recover fragmented DNA between 150 and 200 bp. We used five PCR cycles for the pre-capture and eight PCR cycles for post-capture amplifications. In order to make the capture step cost-efficient, we added unique indices to each library prior to exome capture (pre-capture indexing method), pooled equal amounts of DNA from 16 libraries and then performed the capture. A capture requiring a total of 750 ng indexed DNA, we pooled 46.875 ng of each of the 16 libraries. To minimize uneven capture of each samples, we pooled libraries from samples showing similar quantities of schistosome genomes previously estimated by qPCR. The capture was performed using sets of baits from the SureSelectXT Target Enrichment System (Agilent, Lexington, Massachussetts, USA) (120 bp RNA molecules) specific to each species.
The design of the baits used to capture the S. mansoni exome (SureSelect design ID: S0398493) has already been described in Chevalier et al. (Reference Chevalier2014). We use our experience with the S. mansoni bait design to improve our design strategy for S. haematobium. Unlike the S. mansoni design, (i) we included all exons that passed the low complexity threshold (rather than only the exons to which sequences can be unambiguously aligned); (ii) we included exons from vaccine candidates (Supplementary File 2); and (iii) we designed baits to ensure effective capture of ShSULT-OR gene (Sha_104171), a drug target of particular interest (Taylor et al., Reference Taylor2017). The design (SureSelect design ID: S0742423) was made using the latest S. haematobium genome version (SchHae_1.0; assembly accession number: GCA_000699445.1) and its corresponding annotation. The final set of S. haematobium baits included 156 004 from the nuclear genome and 67 from the mitochondrial genome. These cover 96% (62 106/64 642) of the exons and account for 94% of the exome length (15 002 706 bp/15 895 612 bp). The sequences covered by baits are referred to as the bait regions. Each captured exon was covered by 2.59 baits on average.
We sequenced the pooled barcoded exome libraries using 100 bp pair-end reads (16 samples per lane of flowcell) on HiSeq 2500 sequencer (Illumina). Raw sequence data have been submitted to the NCBI Sequence Read Archive under accession numbers SRP136210 and SRP136277.
Sequencing data analyses
We aligned the sequencing data against the S. mansoni reference genome (v5; ftp://ftp.sanger.ac.uk/pub/pathogens/Schistosoma/mansoni/genome/Assembly-v5/ARCHIVE/sma_v5.0.chr.fa.gz) or the S. haematobium reference genome (SchHae_1.0; assembly accession number: GCA_000699445.1) using BWA (v0.7.12) (Li and Durbin, Reference Li and Durbin2009) and SAMtools (v1.2) (Li et al., Reference Li2009). Realignment around indels was performed using GATK (v3.3-0-g37228af) (McKenna et al., Reference McKenna2010; DePristo et al., Reference DePristo2011). PCR duplicates were marked using picard (v1.136). Q-score recalibration and variant (SNP/indel) calling by the UnifiedGenotyper module were performed using GATK. Bait representation, capture efficiency and read depth analyses were performed using BEDTools (v2.21.0) (Quinlan and Hall, Reference Quinlan and Hall2010). The variant calling set was filtered using VCFtools (v0.1.14) (Danecek et al., Reference Danecek2011) to retain only variants in the bait regions and with a minimum read depth of 10 and a minimum genotype quality score of 80.
Statistics and data representation
Statistics analyses, calculation of capture efficiency, calculation of the read depth ratio of sex chromosome, analysis of read depth surrounding bait regions and data visualizations were performed using R (v3.3.1). Figure 1 summarizes our pipeline for generating and analysing exome sequence from single miracidia.
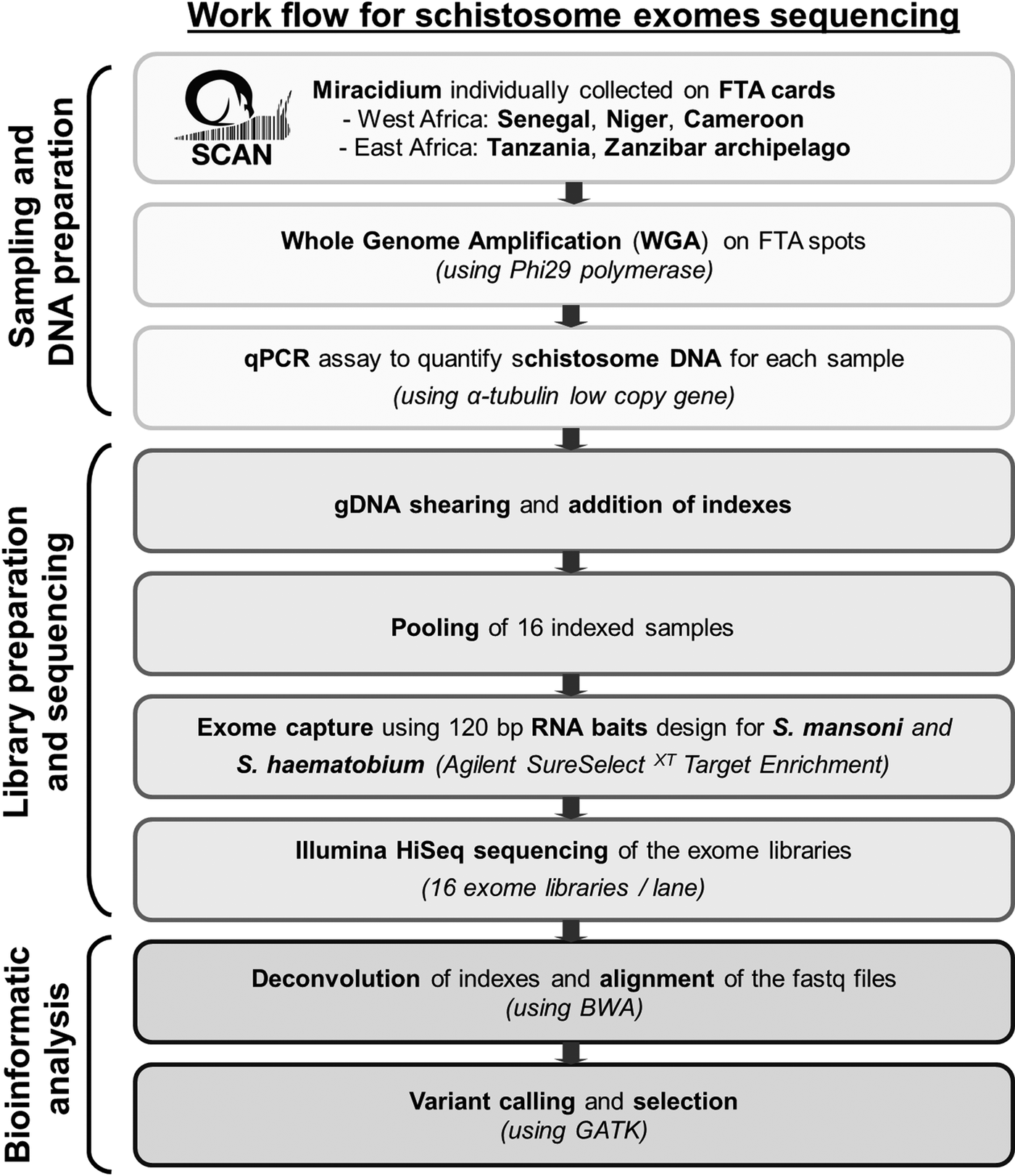
Fig. 1. Workflow summarizing the protocol used to generate and analyse genomic data starting with field collecting miracidium.
Results
WGA of single miracidia
We performed WGA on a total of 412 FTA-preserved samples. These include 101 S. mansoni samples from East Africa (Tanzania) and 90 S. mansoni samples from West Africa (Senegal, Niger and Cameroon) (1–3 samples per patient) as well as 138 S. haematobium samples from East Africa (Zanzibar archipelago) and 83 FTA preserved S. haematobium samples from West Africa (Niger) (1–4 samples per patient). This resulted in an average of 3.8 ± 0.66 µg of DNA (mean ± s.d.) from each S. mansoni sample and an average of 2.8 ± 0.48 µg of DNA from each S. haematobium sample.
The amount of schistosome DNA per sample is critical for downstream genomic assays: samples with no schistosome DNA need to be removed, while samples with low levels (<100 copies) of schistosome template DNA result in failed exome capture. To estimate the quantity of schistosome DNA in each WGA, we quantified a low copy gene (sat-1 for S. mansoni and α-tubulin 2 for S. haematobium) using qPCR. While DNA was successfully amplified from almost 100% of the spots (Table 1), the qPCR assay revealed that only 59.4 and 41.11% of the whole genome amplified samples were positive for S. mansoni from the East and West African samples, respectively. For the S. haematobium samples, 47.10% of schistosome-positive WGA samples were from the East and 66.26% from the West African samples. We found no S. mansoni-positive samples among 14 FTA punches examined from Cameroon (Table 1).
Table 1. Whole genome amplification and qPCR assay summary statistics

Summary of the number and percentage of positive schistosome samples, the number of patients from which schistosome-positive WGA were obtained, the average number of spots tested per patients in order to get at least one positive for schistosome, and the average ( + s.d.) DNA concentration recovered after WGA in ng µL−1 in 130 µL of sample.
a We initially conducted WGA from a single miracidia per patient: further WGA were done only if the initial sample was schistosome-negative by qPCR.
b Only shown for schistosome-positive samples.
The quantity of S. mansoni DNA obtained from single miracidia after WGA varied over five orders of magnitude (Fig. 2) among the 97 S. mansoni-positive samples from East (n = 60) and West Africa (n = 37). Two samples from each region (3% of East African and 5% West African samples) contained <100 copies. WGA material from East African samples showed higher quantities of S. mansoni DNA than WGA material from West African samples (Fig. 2; Wilcoxon test; P = 1.463 × 10−6).
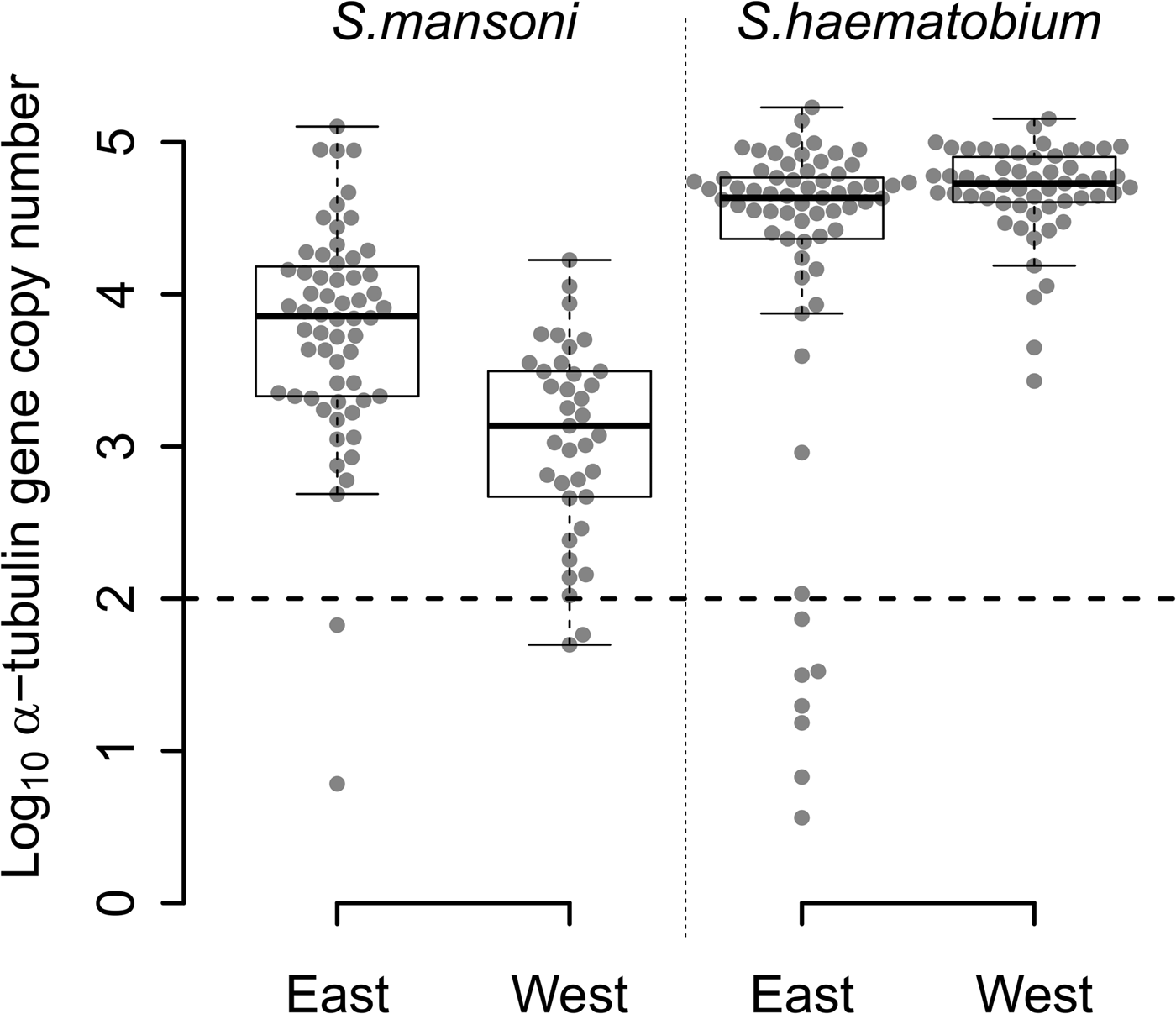
Fig. 2. Distribution of the number of α-tubulin copy among all the WGA schistosome samples tested. Log10 distribution of the number of α-tubulin copy in 20 ng of total DNA, among the 60 East African (Tanzania) and the 37 West African (Senegal and Niger) Schistosoma mansoni samples, and the 65 East African (Zanzibar) and 55 West African (Niger) Schistosoma haematobium samples from FTA-preserved samples. S chistosome mansoni and S. haematobium DNA varies in concentration by three to five orders of magnitude within WGA products. The 11 outliers, below the dotted line threshold, exhibited a very low number of α-tubulin copies (<100 copies in 20 ng of total DNA) and were removed from the next generation sequencing sample set.
We observed similar heterogeneity in schistosome DNA quantity for WGA from the 65 S. haematobium-positive samples from East Africa, while the quantity of DNA from 55 West African S. haematobium WGAs varied by only three orders of magnitude, with no outliers. Only 11% (7/65) of S. haematobium-positive samples, all from East Africa, showed <100 copies. We found a marginally higher quantity of S. haematobium DNA in West compared with East African samples (Fig. 2; Wilcoxon test; P = 0.017).
WGA from S. haematobium samples contained more schistosome DNA than from S. mansoni samples in both East (Fig. 2; Wilcoxon test; P = 1.980 × 10−7) and West African samples (Fig. 2; Wilcoxon test; P = 2.472 × 10−15).
Exome sequencing of single miracidia
We show exome sequencing data from 8 S. mansoni miracidia from a single Tanzanian village (Kigongo, Supplementary File 1) and from eight S. haematobium miracidia from three villages on Pemba island (Zanzibar archipelago: Chambani, Ngwachani and Chanjamjamiri; Supplementary File 1). We observed efficient exome capture and sequencing for both species (Table 2). Sequencing of the exome capture libraries generated from six to 26 million reads for S. mansoni and from five to 18 million reads for S. haematobium (Table 2). A very high proportion of these reads were mapped to their respective genomes (95.5 and 95% on average, respectively) with only one sample from each species showing <90% of mapped reads (84 and 89%, respectively). The remaining DNA sequences, which were not mappable, could either be highly divergent schistosome sequences or more likely human or microbial contaminants present in the WGA sample and captured with the schistosome DNA.
Table 2. Exome capture library statistics

The ratio Z/W is the ratio of the read depth of the Z-linked regions (between 3.5–19.5 and 23.5–31 Mb) over the read depth of the rest of the Z chromosome. We considered a ratio between 0.5 and 0.75 corresponding to females, and a ratio over 0.75 to males. ND: not determined because the Schistosoma haematobium sexual chromosome is not identified in the current assembly.
We captured more than 99% of the bait regions and this was consistent across all eight libraries of each species (Table 2). We obtained an average of 51.74 mean (30.62 median) read depth in the bait regions for the S. mansoni libraries and an average of 38.07 mean (30.62 median) read depth for the S. haematobium libraries (Table 2). Read depth was even across the exome (Fig. 3), except in parts of the Z chromosome in S. mansoni females where read depth is halved (females are ZW). These genome regions (position 3.5–19.5 and 23.5–31 Mb for S. mansoni (Lepesant et al., Reference Lepesant2012)) correspond to the non-recombining heterochromatin domain of the W chromosome. The read depth in the Z-linked region allows us to determine the parasite sex in silico using a read depth ratio between the Z-linked region and the rest of the chromosome. Among the samples present in Table 2, we obtained an equal sex ratio. The S. haematobium genome assembly is poorly resolved and the Z-linked region still remains undefined (Table 2, Fig. 3).

Fig. 3. Read depth of the bait regions for Schistosoma mansoni and Schistosoma haematobium single miracidium libraries. Schistosoma mansoni (Sm) plots show read depth of bait regions on the assembled chromosomes (grey), on unplaced scaffolds that have been assigned to chromosomes or unassigned scaffolds (black). Schistosoma haematobium (Sh) plots show read depth of the concatenated bait regions (bp, base pair).
We also captured genome regions outside the bait regions, as shown previously (Chevalier et al., Reference Chevalier2014). The read depth declines with distance from the bait regions as expected (Table 2). When we included data up to 250 bp around the bait regions, the mean and the median read depth reached ~72 and ~37% of the initial bait regions, respectively. These observations were similar for both S. mansoni and S. haematobium. Therefore, our exome capture provides adequate sequence read depths for at least 250 bp surrounding bait regions allowing us to obtain information regarding adjacent genome regions containing promoters, transcription binding sites and other features of interest.
We were able to robustly identify variable sites in the exome capture libraries from each species (Table 3). The raw variant calling revealed more variable sites in S. mansoni than in S. haematobium. The raw data were further filtered using very stringent parameters (minimum read depth of 10 and genotype quality of 80). After filtering, we retained informative sites accounting for around 74% of the initial sites identified. Among them, the vast majority (96%) were biallelic sites, <1% was multiallelic sites and between 3 and 4% were identified as indel sites.
Table 3. Variant calling statistics

Number of variable sites identified in the eight exome capture libraries of S. mansoni and S. haematobium. We used conservative filtering parameters: minimum read depth of 10 and a minimum genotype quality (GQ) of 80.
Discussion
Utility of WGA and qPCR quality check for FTA-preserved miracidia
WGA provides a very effective approach to generating large amounts of DNA from single miracidia for genome wide analyses (Valentim et al., Reference Valentim2009; Shortt et al., Reference Shortt2017). This approach has previously been used to generate micrograms of DNA from a S. mansoni laboratory genetic cross with a minimal error rate (0.45%) induced by the WGA step (Valentim et al., Reference Valentim2009). The current study extends this approach to WGA of archived miracidia collected in the field. Such material is more challenging compared with laboratory prepared miracidia because the miracidia on FTA cards may contain extensive environmental contamination, such as fecal bacteria or human DNA. Two features of the approach used are worth emphasising. First, the WGA method uses 2 mm FTA punches directly immersed in the WGA reaction, eliminating the need for initial DNA preparation. This produced around 3 µg of material that is sufficient for next generation sequencing applications that require at least 500 ng to 1 µg of DNA. Second, we developed a simple qPCR assay for quantifying amounts of S. mansoni and S. haematobium DNA present in the WGA material. This allows identification of schistosome-positive samples and quantification of schistosome DNA, which is a critical step for the identification of WGA samples suitable for library preparation and for grouping samples in pools with similar amounts of schistosome DNA to minimize variation in read depth.
We observed variation in the numbers of positive schistosome WGAs, ranging from 46.42 to 66.26% for S. mansoni or S. haematobium populations, respectively. These results most likely reflect variations in the miracidia collection protocol (variation in egg washing, in spot sizes on FTA cards) and/or in the training of people collecting miracidia. We observed higher and more uniform concentration of S. haematobium DNA in WGA material, when compared with S. mansoni DNA (Fig. 2). The higher variation in S. mansoni DNA amount is consistent with fecal contamination, as less contamination is expected in S. haematobium isolated from urine. We recommend that future miracidia collections, specifically for genomic work, should wash S. mansoni miracidia in clean water, prior to pipetting onto FTA cards, minimalizing contamination.
Among schistosome-positive samples, we removed those showing low quantity of schistosome DNA (<100 copies of tubulin gene after WGA) because only higher schistosome DNA quantities gave good sequencing data.
Efficient exome capture from FTA preserved miracidia
Preparation of exome capture libraries using WGA material from either S. mansoni or S. haematobium led to high read depth exome sequencing data despite five orders of magnitude variation in the quantity of schistosome DNA present in the WGAs. Using very conservative calling parameters, we scored 85 133 variable sites in S. mansoni and 60 193 in S. haematobium from just eight individual miracidia of each species, which is equivalent to one variant every 166 bp for S. mansoni and every 249 bp for S. haematobium in the bait regions sequenced. This provides a rich source of variation for population exomic analyses and will be the focus of future studies with large numbers of samples.
Our exome capture method has several useful characteristics for working with field-collected miracidia. Exome capture effectively pulls out schistosome DNA (exons) from contaminating human or bacterial DNA so we can minimize the amount of contaminant sequence generated. Capture is very efficient and consistent (more than 99% of the expected region is captured for each sample) so we can sequence to high read depth to generate robust variant calls, or use read depth to estimate copy number. Here, for example, we can use read depth information from the S. mansoni Z-linked sex chromosomes for in silico sexing. The main disadvantages of exome sequencing are some highly variable genes may be poorly captured and many non-coding regions important for gene regulation are not captured (Biesecker et al., Reference Biesecker, Shianna and Mullikin2011), although we do effectively capture non-coding regions adjacent to bait sequences. We note that whole genome sequencing can also be done using WGA miracidia, if precautions are made to minimize contaminations during collection of miracidia.
Costs of exome sequencing miracidia
Exome capture methods are more expensive than other reduced representation library preparation approaches, such as RADseq (Harvey et al., Reference Harvey2016). However, exome capture targets specific genome regions of interest and in a consistent manner for each sample, and shows lower levels of genotyping error and allelic drop out (Attard et al., Reference Attard, Beheregaray and Möller2017) than RADseq and related methods, so it has several advantages. The baits are by far the most expensive part of this method (~US$900 for a given capture) but the pre-capture indexing method allows pooling of up to 16 samples prior the capture. This brings the capture cost down to $56.25 per sample, while the other reagents required for library preparation are $83.75 per sample. The overall cost per sample is therefore approximately $140. The cost of a flow cell lane for a HiSeq 2500, sufficient to sequence 16 libraries, is currently around $2000 in our facility, so the final cost is $265 per sample for an average read depth of 50. To achieve the same read depth when sequencing the entire schistosome genome requires two samples per lane, costing around $1000 per sample (library preparation cost is, in this case, negligible). Capturing and sequencing exomes also reduces both the analysis time and the storage capacity required for housing data because the exome represents just 4% of the complete genome (15 vs 365 Mb genome). Exome capture is currently about 4-fold less expensive than whole genome sequencing at present, ignoring the additional costs for data storage and analysis of whole genome data. The central advantage of exome capture over both RADseq and whole genome sequencing is that contaminating sequences can be effectively removed.
Our established workflow using the WGA method and qPCR quality check (Fig. 1) will facilitate the transition from population genetics using a handful of loci, to population genomics using genome-wide information. Using these tools, we will be able to fully exploit large collections of archived miracidia, such as those available through SCAN. For example, analysis of archived miracidia collected may be particularly useful for retrospective analyses of loci underlying drug resistance (Chevalier et al., Reference Chevalier2016). This same approach can also be used for microscopic larval stages of other parasite species.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018000811
Acknowledgements
We gratefully acknowledge the help and support of our collaborator in Senegal, the late Oumar T. Diaw.
Financial support
Work at Texas Biomedical Research Institute (TBRI) was supported by the National Institute of Allergy and Infectious Diseases Grants (R01 AI097576-01 and 5R21AI096277-01), by the National Center for Advancing Translational Sciences (CTSA/IIMS award no. UL1TR001120) and by Texas Biomedical Research Institute Forum grant (award 0467) and was conducted in facilities constructed with support from Research Facilities Improvement Program grant C06 RR013556 and RR017515 from the National Center for Research Resources of the National Institutes of Health. WL was supported by a Cowles Fellowship. CONTRAST was funded by the European Commission (FP6 STREP contract no: 032203). SCORE (https://score.uga.edu/) collecting activities in Tanzania and Zanzibar were funded by the University of Georgia Research Foundation Inc. (prime award no. 50816, sub awards RR374-053/4785416 and RR374-053/4893206), which is funded by the Bill & Melinda Gates Foundation for SCORE projects. SCAN is funded with support from the Wellcome Trust (grant no. 104958/Z/14/Z).
Conflict of interest
None.









 Open access
Open access