


INTRODUCTION
Malaria is one the most serious infectious diseases of humans with over 250 million clinical cases every year worldwide. The infection is transmitted to humans by Anopheles mosquitoes that are infected with parasites of the genus Plasmodium. Most cases of severe disease are caused by Plasmodium falciparum, which is endemic in sub-Saharan Africa and throughout the tropics. The blood stage of the parasite is largely responsible for malaria-associated pathology (Miller et al. Reference Miller, Baruch, Marsh and Doumbo2002). The fatalities are associated with a range of various disease syndromes including acute respiratory distress, metabolic acidosis, hypoglycaemia, renal failure, anaemia, pulmonary oedema and cerebral malaria (CM) (White and Ho, Reference White and Ho1992). This disease syndrome is the most severe complication resulting from P. falciparum infection and accounts for nearly 1 million deaths every year (Murray et al. Reference Murray, Rosenfeld, Lim, Andrews, Foreman, Haring, Fullman, Naghavi, Lozano and Lopez2012). Children under the age of five are particularly susceptible to this condition, which is characterized by convulsions, seizures and coma. Similar to young children, pregnant women (particularly primigravid women) are at an increased risk of infection and might develop placental malaria (PM). This serious complication has been found to be associated with adverse pregnancy outcomes including premature labour, intrauterine growth retardation and low birth weight delivery, and is responsible for more than 75 000 infant deaths each year (McGregor, Reference McGregor1984; Steketee et al. Reference Steketee, Nahlen, Parise and Menendez2001; Desai et al. Reference Desai, ter Kuile, Nosten, McGready, Asamoa, Brabin and Newman2007).
Mature forms of blood-stage malaria express parasitic proteins on the surface of the infected erythrocyte (such as P. falciparum Erythrocyte Membrane Protein 1), which allows them to bind to endothelial cells, sequester in vascular beds and avoid clearance in the spleen. Although the precise mechanisms leading to severe malaria syndromes are not completely understood, it is accepted that sequestration of parasitized red blood cells (pRBC) is a major determinant of disease development. Parasite sequestration is thought to induce obstructions in blood flow resulting in hypoxia and haemorrhages (Miller et al. Reference Miller, Baruch, Marsh and Doumbo2002) that appear to be associated with the development of organ-specific syndromes such CM and PM. A large body of literature indicates that in addition to parasite sequestration, inflammatory responses mediated by cytokines such as TNF (Molyneux et al. Reference Molyneux, Engelmann, Taylor, Wirima, Aderka, Wallach and Grau1993), IFN-γ and IL-1β (Pongponratn et al. Reference Pongponratn, Turner, Day, Phu, Simpson, Stepniewska, Mai, Viriyavejakul, Looareesuwan, Hien, Ferguson and White2003) are associated with disease severity in human malaria.
Much useful evidence on the inflammatory mechanisms contributing to the induction of severe malaria has been provided by the Plasmodium berghei ANKA model. This rodent infection has many features in common with human disease and is thus the best available model for certain aspects of clinical malaria (Schofield and Grau, Reference Schofield and Grau2005; Hansen, Reference Hansen2012). Like in humans, P. berghei-ANKA pRBC have been found to accumulate in brains of susceptible mice during infection. Moreover, recent evidence revealed that P. berghei-ANKA pRBC adhere to brain microvascular endothelial cells in a VCAM-1-dependent fashion (El-Assaad et al. Reference El-Assaad, Wheway, Mitchell, Lou, Hunt, Combes and Grau2013). A large body of work demonstrated that host immune responses elicited during P. berghei ANKA infection result in detrimental inflammation and contribute to cerebral disease induction. Host responses mediated by inflammatory cytokines such as TNF (Grau et al. Reference Grau, Fajardo, Piguet, Allet, Lambert and Vassalli1987), LT-α (Engwerda et al. Reference Engwerda, Mynott, Sawhney, De Souza, Bickle and Kaye2002), IFN-γ (Grau et al. Reference Grau, Heremans, Piguet, Pointaire, Lambert, Billiau and Vassalli1989) and effector cells such as CD4+ (Grau et al. Reference Grau, Piguet, Engers, Louis, Vassalli and Lambert1986; Yanez et al. Reference Yanez, Manning, Cooley, Weidanz and van der Heyde1996; Villegas-Mendez et al. Reference Villegas-Mendez, Greig, Shaw, de Souza, Gwyer Findlay, Stumhofer, Hafalla, Blount, Hunter, Riley and Couper2012) and CD8+ T cells (Belnoue et al. Reference Belnoue, Kayibanda, Vigario, Deschemin, van Rooijen, Viguier, Snounou and Renia2002; Nitcheu et al. Reference Nitcheu, Bonduelle, Combadiere, Tefit, Seilhean, Mazier and Combadiere2003), NKT cells (Hansen et al. Reference Hansen, Siomos, Buckingham, Scalzo and Schofield2003) and NK cells (Hansen et al. Reference Hansen, Bernard, Nie and Schofield2007; Ryg-Cornejo et al. Reference Ryg-Cornejo, Nie, Bernard, Lundie, Evans, Crabb, Schofield and Hansen2013) have been shown to contribute to severe malaria in this model.
In addition to pRBC, human post-mortem studies revealed the presence of leucocytes and platelets within the brain microvasculature in a substantial proportion of CM cases (Porta et al. Reference Porta, Carota, Pizzolato, Wildi, Widmer, Margairaz and Grau1993; Patnaik et al. Reference Patnaik, Das, Mishra, Mohanty, Satpathy and Mohanty1994; Grau et al. Reference Grau, Mackenzie, Carr, Redard, Pizzolato, Allasia, Cataldo, Taylor and Molyneux2003; Hunt and Grau, Reference Hunt and Grau2003; Taylor et al. Reference Taylor, Fu, Carr, Whitten, Mueller, Fosiko, Lewallen, Liomba and Molyneux2004). Interestingly, sequestered monocytes and macrophages were found to be more abundant in CM paediatric patients than in severe malarial anaemia (SMA) patients (Coltel et al. Reference Coltel, Combes, Hunt and Grau2004). Similarly, although the sequestration of pRBC in the placenta appears to be responsible for initiation of pathology, histological evidence revealed the infiltration of various leucocyte populations including monocytes, macrophages, T cells and granulocytes (Ordi et al. Reference Ordi, Menendez, Ismail, Ventura, Palacin, Kahigwa, Ferrer, Cardesa and Alonso2001). An important intervillositis occurs in PM cases with evident sequestration of both pRBC and leucocytes preventing efficient blood flow across the placenta. Moreover, mononuclear intervillous inflammatory infiltration has been found to be associated with low birth weight and preterm delivery (Ordi et al. Reference Ordi, Ismail, Ventura, Kahigwa, Hirt, Cardesa, Alonso and Menendez1998, Reference Ordi, Menendez, Ismail, Ventura, Palacin, Kahigwa, Ferrer, Cardesa and Alonso2001).
In experimental mouse models, several leucocyte populations including macrophages, neutrophils, T cells, NK cells and platelets have been found in brain blood vessels of CM-affected mice during infection (Grau et al. Reference Grau, Fajardo, Piguet, Allet, Lambert and Vassalli1987, Reference Grau, Tacchini-Cottier, Vesin, Milon, Lou, Piguet and Juillard1993; Ma et al. Reference Ma, Hunt, Madigan and ChanLing1996; Belnoue et al. Reference Belnoue, Kayibanda, Vigario, Deschemin, van Rooijen, Viguier, Snounou and Renia2002; Nitcheu et al. Reference Nitcheu, Bonduelle, Combadiere, Tefit, Seilhean, Mazier and Combadiere2003; Hansen et al. Reference Hansen, Bernard, Nie and Schofield2007; Lundie et al. Reference Lundie, de Koning-Ward, Davey, Nie, Hansen, Lau, Mintern, Belz, Schofield, Carbone, Villadangos, Crabb and Heath2008). From these populations, CD8+ T cells comprise a high proportion of the brain-sequestered leucocyte pool of P. berghei-ANKA-infected mice and appear to mediate CM via a perforin-dependent mechanism (Belnoue et al. Reference Belnoue, Kayibanda, Vigario, Deschemin, van Rooijen, Viguier, Snounou and Renia2002; Nitcheu et al. Reference Nitcheu, Bonduelle, Combadiere, Tefit, Seilhean, Mazier and Combadiere2003). Moreover, infection of β 2-microglobulin−/− mice as well as antibody depletion studies demonstrated that CD8+ T cells contribute to the induction of experimental cerebral malaria (ECM) (Yanez et al. Reference Yanez, Manning, Cooley, Weidanz and van der Heyde1996).
The increasing body of evidence illustrating the presence of sequestered host cells together with infected erythrocytes in organs such as the brain or placenta during infection suggested that in addition to secreting cytokines and mediating systemic responses, leucocytes might also contribute to disease by migrating to the site of parasite sequestration, thereby exacerbating organ-specific inflammation. These observations attracted substantial interest in identifying trafficking pathways by which leucocytes are recruited to target organs during severe malaria syndromes and the potential contribution of chemokines to disease. In this review we have summarized the main findings to date investigating the role of chemokines during severe malaria including experimental infection in murine models as well as human field studies. The implications of those findings for the induction of pathogenesis and immunity to malaria are discussed.
THE ROLE OF CHEMOKINES IN HOMOEOSTASIS AND INFLAMMATION
Chemoattractant cytokines or chemokines are key regulators of leucocyte trafficking. Chemokine-guided movement allows leucocyte migration to various lymphoid tissues and deployment of immune cells to peripheral sites of pathogen challenge and inflammation. Chemokines are a superfamily of low molecular weight polypeptides of about 8–14 kDa, which bind and signal through seven-transmembrane spanning G-protein coupled receptors (GPCRs). To date, nearly 50 chemokines have been identified (Table 1) as well as numerous chemokine-binding proteins that do not induce cell-signalling events (Murphy, Reference Murphy2002). Different chemokines share a significant degree of sequence homology and are subdivided into CC, CXC, CX3C and C subfamilies based on the relative positions of conserved cysteine residues near the N-terminus (Luster, Reference Luster1998).
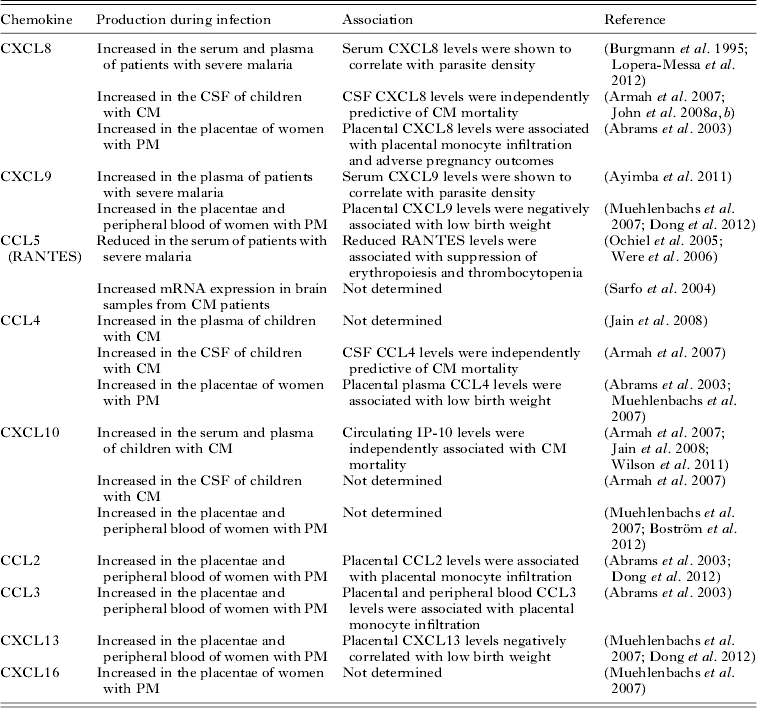
Table 1. Chemokines and chemokine receptors

a Chemokines are identified with both common names and systematic names in parenthesis. Bold font identifies inflammatory/inducible chemokines. Homoeostatic chemokines are shown in italic font. Bold and italic font denotes chemokines belonging to both classifications.
Chemokines and chemokine receptors are essential in two distinct processes of leucocyte migration. The first is the directional migration and positioning of leucocytes within lymphoid organs as well as in peripheral tissues. In this context, cells encounter chemoattractant signals in a complex spatial and temporal pattern, in which cells follow a gradient of increasing concentration towards the source of the chemokine (reviewed in Rot and von Andrian (Reference Rot and von Andrian2004). The second trafficking process mediated by chemokines involves the arrest of migrating leucocytes on the vascular endothelium followed by extravasation from the blood vessel into lymphoid and inflamed tissues. In this process, chemokines are immobilized by sulphated sugars of the glycosaminoglycan (GAG) family on the luminal surface of vascular endothelial cells, which is essential for optimal adhesion of leucocytes to the endothelium (Rot, Reference Rot1992; Tanaka et al. Reference Tanaka, Adams and Shaw1993).
Functionally, chemokines can be divided into two broad categories: constitutively expressed chemokines that maintain homoeostatic functions, and inducible chemokines that are usually upregulated in response to infection and/or inflammation (Sallusto et al. Reference Sallusto, Mackay and Lanzavecchia2000; Moser et al. Reference Moser, Wolf, Walz and Loetscher2004). This distinction can be somewhat arbitrary as some chemokines fall into both categories depending on the biological context (Table 1). Inflammatory chemokines are produced by several cell types including stromal, endothelial and epithelial cells as well as leucocytes. Constitutive chemokines are produced in the thymus and lymphoid tissues and regulate homoeostatic functions of the immune system such as maintaining steady-state leucocyte homing and cell compartmentalization within lymphoid organs (Yoshie et al. Reference Yoshie, Imai and Nomiyama1997). Leucocytes have the capacity to switch the expression of receptors from constitutive to inflammatory chemokines, which allows for different migratory patterns. For example, upon antigen-specific activation and differentiation into effector function, T cells upregulate a repertoire of chemokine receptors and adhesion molecules, which facilitates their exit from secondary lymphoid organs and migration to peripheral inflamed tissues (Xie et al. Reference Xie, Lim, Luscinskas and Lichtman1999; Weninger et al. Reference Weninger, Crowley, Manjunath and von Andrian2001).
Different effector cell subsets have been shown to express distinct chemokine receptor patterns, allowing them specific migration patterns to exert their function. Polarization of CD4+ T cells to TH1 and TH2 subsets is associated with the upregulation of a distinct set of chemokines and their chemokine receptors, which is influenced by the cytokine milieu during priming (Sallusto et al. Reference Sallusto, Lanzavecchia and Mackay1998; Bromley et al. Reference Bromley, Mempel and Luster2008). Typically, TH1 cells are predominantly characterized by the expression of CXCR3, CCR5 and CXCR6, whereas TH2 cells express CCR3, CCR4 and CCR8. However, the association of chemokine receptors with T-helper phenotypes in vivo shows a much more complex profile (Annunziato et al. Reference Annunziato, Cosmi, Galli, Beltrame, Romagnani, Manetti, Romagnani and Maggi1999; Kim et al. Reference Kim, Rott, Kunkel, Genovese, Andrew, Wu and Butcher2001), suggesting that CD4+ T cell subsets cannot always be accurately identified solely on the basis of their chemokine receptor expression. Moreover, increasing evidence suggests that the chemokine receptor repertoire expressed on activated T cells shows a degree of dynamic plasticity, whereby several factors including strength of antigenic signals, tissue-specific imprinting by priming dendritic cells or cytokine milieu during priming can influence the trafficking phenotype of T cells (reviewed in Mora and von Andrian (Reference Mora and von Andrian2006).
ASSOCIATION OF CHEMOKINES WITH HUMAN SEVERE MALARIA SYNDROMES
Several inflammatory chemokines have been found to be associated with severe malaria syndromes in case-control studies (Table 2). Increased levels of CXCL8 and CXCL9 as well as reduced levels of CCL5 (also known as RANTES) have been observed in severe malaria patients (Burgmann et al. Reference Burgmann, Hollenstein, Wenisch, Thalhammer, Looareesuwan and Graninger1995; Ochiel et al. Reference Ochiel, Awandare, Keller, Hittner, Kremsner, Weinberg and Perkins2005; Ayimba et al. Reference Ayimba, Hegewald, Segbena, Gantin, Lechner, Agosssou, Banla and Soboslay2011; Lopera-Mesa et al. Reference Lopera-Mesa, Mita-Mendoza, van de Hoef, Doumbia, Konate, Doumbouya, Gu, Traore, Diakite, Remaley, Anderson, Rodriguez, Fay, Long, Diakite and Fairhurst2012). Moreover, CXCL8 and CXCL9 levels have been shown to correlate with parasite density (Burgmann et al. Reference Burgmann, Hollenstein, Wenisch, Thalhammer, Looareesuwan and Graninger1995; Ayimba et al. Reference Ayimba, Hegewald, Segbena, Gantin, Lechner, Agosssou, Banla and Soboslay2011) suggesting that increased parasitic loads might induce these responses. In support of that concept, in vitro studies demonstrated that P. falciparum-infected erythrocytes are able to induce CCL2, CCL20, CXCL1, CXCL2, CXCL6 and CXCL8 production by human endothelial cells (Viebig et al. Reference Viebig, Wulbrand, Forster, Andrews, Lanzer and Knolle2005; Chakravorty et al. Reference Chakravorty, Carret, Nash, Ivens, Szestak and Craig2007; Tripathi et al. Reference Tripathi, Sha, Shulaev, Stins and Sullivan2009).
Table 2. Association between chemokines and the outcome of human malaria infections
Severe malarial anaemia is one of the most common clinical manifestations of disease in children (Marsh et al. Reference Marsh, Forster, Waruiru, Mwangi, Winstanley, Marsh, Newton, Winstanley, Warn, Peshu, Pasvol and Snow1995). Both erythropoietic suppression (Kurtzhals et al. Reference Kurtzhals, Rodrigues, Addae, Commey, Nkrumah and Hviid1997) and RBC destruction (Looareesuwan et al. Reference Looareesuwan, Davis, Pukrittayakamee, Supanaranond, Desakorn, Silamut, Krishna, Boonamrung and White1991) are thought to contribute to the development of SMA. Reduced circulating levels of CCL5 have been found to be associated with SMA in children (Ochiel et al. Reference Ochiel, Awandare, Keller, Hittner, Kremsner, Weinberg and Perkins2005; Were et al. Reference Were, Ouma, Otieno, Orago, Ong'echa, Vulule, Keller and Perkins2006). Moreover, reduced CCL5 production was associated with the suppression of erythropoiesis and malaria-induced thrombocytopaenia (Were et al. Reference Were, Ouma, Otieno, Orago, Ong'echa, Vulule, Keller and Perkins2006), suggesting that CCL5 may be involved in the regulation of the erythorpoietic response during malaria infection. It is thought that monocytic-acquisition of P. falciparum hemozoin (pfHz) may in part contribute to CCL5 suppression during SMA, as children with high levels of intramonocytic pfHz have been shown to have lower circulating CCL5 levels than children with only low levels of intramonocytic pfHz (Were et al. Reference Were, Davenport, Yamo, Hittner, Awandare, Otieno, Ouma, Orago, Vulule, Ong'echa and Perkins2009). This is further supported by the observation that CCL5 production in IFN-γ-stimulated PBMCs from malaria-infected children decreased with increasing levels of intramonocytic pfHZ (Were et al. Reference Were, Davenport, Yamo, Hittner, Awandare, Otieno, Ouma, Orago, Vulule, Ong'echa and Perkins2009).
Although reduced circulating levels of CCL5 have been shown to be associated with CM (John et al. Reference John, Opika-Opoka, Byarugaba, Idro and Boivin2006), increased transcription of this chemokine has been observed in brain samples from CM patients (Sarfo et al. Reference Sarfo, Singh, Lillard, Quarshie, Gyasi, Armah, Adjei, Jolly and Stiles2004). Circulating levels of CCL4 (also known as MIP-1β), CXCL10 (also known as IP-10), CXCL4 and CXCL8 have been found to be significantly elevated in CM cases compared with mild malaria cases or healthy control individuals (John et al. Reference John, Opika-Opoka, Byarugaba, Idro and Boivin2006, Reference John, Park, Sam-Agudu, Opoka and Bolvin2008b ; Armah et al. Reference Armah, Wilson, Sarfo, Powell, Bond, Anderson, Adjei, Gyasi, Tettey, Wiredu, Tongren, Udhayakumar and Stiles2007; Jain et al. Reference Jain, Armah, Tongren, Ned, Wilson, Crawford, Joel, Singh, Nagpal, Dash, Udhayakumar, Singh and Stiles2008; Wilson et al. Reference Wilson, Jain, Roberts, Lucchi, Joel, Singh, Nagpal, Dash, Udhayakumar, Singh and Stiles2011). Amongst these chemokines, CXCL10 appears to be the most accurate predictor of CM mortality (Jain et al. Reference Jain, Armah, Tongren, Ned, Wilson, Crawford, Joel, Singh, Nagpal, Dash, Udhayakumar, Singh and Stiles2008; Wilson et al. Reference Wilson, Jain, Roberts, Lucchi, Joel, Singh, Nagpal, Dash, Udhayakumar, Singh and Stiles2011). Moreover, cerebrospinal fluid levels of CXCL10, CXCL8 and CCL4 have been found to be significantly higher in children with CM compared with children with SMA and non-malaria-infected individuals (Armah et al. Reference Armah, Wilson, Sarfo, Powell, Bond, Anderson, Adjei, Gyasi, Tettey, Wiredu, Tongren, Udhayakumar and Stiles2007; John et al. Reference John, Panoskaltsis-Mortari, Opoka, Park, Orchard, Jurek, Idro, Byarugaba and Boivin2008a ).
CHEMOKINES AND HUMAN PLACENTAL MALARIA
Most of the available information to date on the role of chemokines in the pathogenesis of severe malaria in humans has been obtained from PM studies. During this serious pregnancy complication, parasite sequestration appears to induce secretion of pro-inflammatory cytokines and chemokines as well as the recruitment of macrophages and monocytes to the intervillous space (Rogerson et al. Reference Rogerson, Brown, Pollina, Abrams, Tadesse, Lema and Molyneux2003a , Reference Rogerson, Pollina, Getachew, Tadesse, Lema and Molyneux b ). Consistent with these observations, increased levels of CCL2 (also known as MCP-1), CCL3 (also known as MIP-1α), CCL4, CXCL8, CXCL9, CXCL13 and CXCL16 have been observed in the placentae of women with PM (Abrams et al. Reference Abrams, Brown, Chensue, Turner, Tadesse, Lema, Molyneux, Rochford, Meshnick and Rogerson2003; Chaisavaneeyakorn et al. Reference Chaisavaneeyakorn, Moore, Mirel, Othoro, Otieno, Chaiyaroj, Shi, Nahlen, Lal and Udhayakumar2003; Muehlenbachs et al. Reference Muehlenbachs, Fried, Lachowitzer, Mutabingwa and Duffy2007; Dong et al. Reference Dong, Kurtis, Pond-Tor, Kabyemela, Duffy and Fried2012). Reduced levels of CCL5 and increased levels of CXCL10, CXCL9 and CCL2 have also been observed in the peripheral blood of women with PM (Bostrom et al. Reference Boström, Ibitokou, Oesterholt, Schmiegelow, Persson, Minja, Lusingu, Lemnge, Fievet, Deloron, Luty and Troye-Blomberg2012; Dong et al. Reference Dong, Kurtis, Pond-Tor, Kabyemela, Duffy and Fried2012). However, only placental CCL4, CXCL8, CXCL9 and CXCL13 have been associated with adverse pregnancy outcomes such as low birth weight deliveries (Abrams et al. Reference Abrams, Brown, Chensue, Turner, Tadesse, Lema, Molyneux, Rochford, Meshnick and Rogerson2003; Muehlenbachs et al. Reference Muehlenbachs, Fried, Lachowitzer, Mutabingwa and Duffy2007; Dong et al. Reference Dong, Kurtis, Pond-Tor, Kabyemela, Duffy and Fried2012).
The β-chemokines CCL2, CCL3 and CCL4 are chemo-attractants for monocytes (Rollins, Reference Rollins1997), while CXCL8 is thought to promote monocyte adhesion (Gerszten et al. Reference Gerszten, Garcia-Zepeda, Lim, Yoshida, Ding, Gimbrone, Luster, Luscinskas and Rosenzweig1999). Thus, production of these chemokines in the placenta during infection may promote monocyte infiltration of the intervillous space. Consistent with this idea, it has been reported that placental CCL2, CCL3 and CXCL8 levels are associated with placental monocyte infiltration (Abrams et al. Reference Abrams, Brown, Chensue, Turner, Tadesse, Lema, Molyneux, Rochford, Meshnick and Rogerson2003). The intravillous infiltrate primarily consists of monocytes and macrophages. Small numbers of B cells, T cells and granulocytes have also been observed in this infiltrate (Ordi et al. Reference Ordi, Menendez, Ismail, Ventura, Palacin, Kahigwa, Ferrer, Cardesa and Alonso2001). Although it is unclear how these cells migrate to the intervillous space during infection, it is possible that recruitment of B and T cells may be mediated by CXCL13 and CXCL9 respectively.
Both maternal and fetal cells appear to contribute to chemokine production in the placenta during PM. Histopathological analysis of malaria-infected placentae has shown that maternal macrophages and fetal stromal cells contribute to CCL3, CCL4 and CXCL13 production in this organ (Abrams et al. Reference Abrams, Brown, Chensue, Turner, Tadesse, Lema, Molyneux, Rochford, Meshnick and Rogerson2003; Muehlenbachs et al. Reference Muehlenbachs, Fried, Lachowitzer, Mutabingwa and Duffy2007), while CCL2, CCL3, CCL4 and CXCL10 have been found in intervillous blood monocular cells isolated from malaria-infected placentae (Suguitan et al. Reference Suguitan, Leke, Fouda, Zhou, Thuita, Metenou, Fogako, Megnekou and Taylor2003). In addition, in vitro differentiated syncytiotrophoblasts have been shown to produce CCL3, CCL4 and CXCL8 in response to stimulation with malaria hemozoin (Lucchi et al. Reference Lucchi, Peterson and Moore2008, Reference Lucchi, Sarr, Owino, Mwalimu, Peterson and Moore2011), which is highly abundant in the placenta during PM (Galbraith et al. Reference Galbraith, Faulk, Galbraith, Holbrook and Bray1980).
THE ROLE OF CHEMOKINES IN SEVERE DISEASE AND IMMUNITY: LESSONS FROM MOUSE INFECTION MODELS
The initial observations supporting a role for inflammatory chemokines in the aetiology of ECM were provided by gene expression studies. Microarray analysis revealed that the expression of CCR5, CXCR3 and CCR1 and several of their chemokines including CXCL9, CXCL10, CCL2, CCL3 and CCL9 significantly increases in response to infection in CM-affected mice (Sexton et al. Reference Sexton, Good, Hansen, D'Ombrain, Buckingham, Simpson and Schofield2004; Hansen et al. Reference Hansen, Evans, D'Ombrain, Bernard, Sexton, Buckingham, Scalzo and Schofield2005). CCL5, CCR1, CCR3 and CCR5 mRNA expression was also detected in brains of Swiss Webster mice and appeared to contribute to the inflammatory response that results in cellular degradation in the cerebellum during Plasmodium yoelii (17XL) infection (Sarfo et al. Reference Sarfo, Armah, Irune, Adjei, Olver, Singh, Lillard and Stiles2005). Further chemokine gene expression studies in brains of ECM-susceptible mice confirmed that CXCL10, CXCL9 and CCL5 and to lesser extent CCL2, CCL7, CCL3, CCL4 and CXCL2 are upregulated during infection with P. berghei ANKA (Hanum et al. Reference Hanum, Hayano and Kojima2003; Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008; Miu et al. Reference Miu, Mitchell, Muller, Carter, Manders, McQuillan, Saunders, Ball, Lu, Campbell and Hunt2008; Van den Steen et al. Reference Van den Steen, Deroost, Aelst, Geurts, Martens, Struyf, Nie, Hansen, Matthys, Damme and Opdenakker2008). Interestingly, the expression of CCL2, CCL5, CCL4, CXCL9 and CXCL10 is induced by either IFN-γ or TNF, which is consistent with the important role of these pro-inflammatory cytokines in ECM pathogenesis.
To study the mechanism of leucocyte recruitment to the brain during malaria infection several studies analysed the chemokine receptor usage of brain-sequestered leucocytes from infected mice. CD8+ T cells isolated from the spleen and brain of malaria-infected mice significantly upregulated CCR5, CCR2, CXCR4 and CXCR3 expression (Nitcheu et al. Reference Nitcheu, Bonduelle, Combadiere, Tefit, Seilhean, Mazier and Combadiere2003), initially suggesting that trafficking via these chemokine receptors could be involved in the recruitment of inflammatory cells during severe disease. However, further studies indicated that CCR2 deficient mice (Table 3) are fully susceptible to ECM (Belnoue et al. Reference Belnoue, Costa, Vigario, Voza, Gonnet, Landau, Van Rooijen, Mack, Kuziel and Renia2003a ). Although there has been conflicting evidence on the role of CCR5 in ECM with CCR5−/− mice reported to be either 80% resistant to P. berghei ANKA-mediated CM (Belnoue et al. Reference Belnoue, Kayibanda, Deschemin, Viguier, Mack, Kuziel and Renia2003b ) or to display only a delayed onset of cerebral disease symptoms (Nitcheu et al. Reference Nitcheu, Bonduelle, Combadiere, Tefit, Seilhean, Mazier and Combadiere2003), mixed-Plasmodium species infection experiments support a role for this trafficking pathway in CM. Plasmodium berghei ANKA-induced CM is inhibited by the co-infection with the non-virulent P. yoelii yoelii 17X clone 1.1 (Table 3). Protection was found to be associated with reduced accumulation of CD8+ T cells in the brain vasculature as well as reduced CCL3, CCL4 and CCL5 levels in the brain (Clark and Phillips, Reference Clark and Phillips2011).
Table 3. Effect of genetic deletion or neutralization of chemokines/chemokine receptors on the outcome of malaria infection in rodent models

Further phenotypic characterization of leucocytes isolated from brain blood vessels of malaria-infected mice by flow cytometry revealed that 80–90% of NK cells and T cells expressed CXCR3, indicating that the expression of this chemokine receptor is strongly associated with lymphocyte trafficking during CM (Hansen et al. Reference Hansen, Bernard, Nie and Schofield2007). Moreover, in vitro chemotaxis assays revealed that whereas T cells from naive mice were unable to migrate in response to the CXCR3-ligand CXCL10, T cells from P. berghei ANKA-infected mice showed a 3-fold increase in their CXCL10-mediated chemotaxis (Hansen et al. Reference Hansen, Bernard, Nie and Schofield2007), indicating that during malaria, T cells acquire the capacity to migrate in response to this chemokine. This appears to be a feature specific of CM-susceptible mouse strains, as T cells from malaria-infected CM-resistant BALB/c animals were found to be unable to respond to CXCL10 chemotactic stimulus (Van den Steen et al. Reference Van den Steen, Deroost, Aelst, Geurts, Martens, Struyf, Nie, Hansen, Matthys, Damme and Opdenakker2008). Together, this evidence supports a role for CXCR3-mediated trafficking in the migration of pathogenic T cells to the brain during ECM. In agreement, it has been found that 70–80% of CXCR3−/− mice are resistant to P. berghei-mediated CM (Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008; Miu et al. Reference Miu, Mitchell, Muller, Carter, Manders, McQuillan, Saunders, Ball, Lu, Campbell and Hunt2008).
The relative involvement of the 3 CXCR3 chemokines (CXCL9, CXCL10 and CXCL11) in the recruitment of inflammatory leucocytes to the brain of malaria-infected mice has been extensively investigated (Table 3). As CXCL11 is not normally expressed in CM-susceptible C57BL/6 mice (usually used in ECM experiments) a role for this chemokine in this model is not expected. CXCL9−/− mice were found to be partially protected from P. berghei-ANKA-mediated CM (Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008). However, whether the increased survival rates to malaria infection in the absence of this chemokine result from reduced leucocyte recruitment to the brain and/or a differential induction of immune response to infection has not been examined.
The role of the CXCR3 chemokine CXCL10 has been studied in more detail. CXCL10−/− mice or mice receiving anti-CXCL10 neutralizing antibodies over the course of P. berghei-ANKA infection were found to have reduced cerebral intravascular inflammation and were protected against fatality (Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009). Moreover, pharmacological inhibition of CXCL10 together with anti-malarial therapy also protects infected animals from CM-mediated mortality (Wilson et al. Reference Wilson, Solomon, Anderson, Patrickson, Pitts, Bond, Liu and Stiles2013). Interestingly, CXCL10 blockade during infection resulted in significantly reduced peripheral parasitaemia, suggesting that the absence of this chemokine has a beneficial effect for the development of parasite-specific responses involved in the control of parasite replication (Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009). Since Plasmodium spp. are blood-borne parasites, the spleen constitutes a key site in the initiation of immune responses and control of parasite replication (Looareesuwan et al. Reference Looareesuwan, Suntharasamai, Webster and Ho1993; Sayles et al. Reference Sayles, Yanez and Wassom1993; Yap and Stevenson, Reference Yap and Stevenson1994; Chotivanich et al. Reference Chotivanich, Udomsangpetch, McGready, Proux, Newton, Pukrittayakamee, Looareesuwan and White2002). Moreover, it has been proposed that the spleen is the site of initial induction of inflammatory cells that migrate to the site of parasite sequestration in target organs (Renia et al. Reference Renia, Potter, Mauduit, Rosa, Kayibanda, Deschemin, Snounou and Gruner2006). Consistent with that concept, the increased resistance to infection observed in the absence of CXCL10-mediated cell trafficking was found to be associated with a preferential accumulation and subsequent expansion of parasite-specific CD4+ T cells in spleens of infected animals (Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009). Overall these findings are consistent with two concepts: (1) trafficking pathways might control the balance between pathogenic organ-specific inflammation and spleen-mediated protective immunity and (2) inflammatory processes that occur during infection are not only detrimental for their involvement in severe disease but can also compromise the induction of anti-parasite immunity by inducing leucocyte migration away from the spleen.
The cellular sources responsible for the secretion of CXCR3 chemokines during ECM have not been extensively investigated, with only a few studies focusing primarily on brain tissue. Gene expression analysis (Miu et al. Reference Miu, Mitchell, Muller, Carter, Manders, McQuillan, Saunders, Ball, Lu, Campbell and Hunt2008) as well as immunohistochemistry approaches (Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008) revealed that CXCL9 is predominantly expressed by endothelial cells and surrounding microglial cells. There is conflicting evidence on the cellular sources of CXCL10 during infection. Whereas CXCL10 RNA was found to be upregulated in brain endothelial cells and astrocytes clustering around IP-10 positive microvessels (Miu et al. Reference Miu, Mitchell, Muller, Carter, Manders, McQuillan, Saunders, Ball, Lu, Campbell and Hunt2008), immunohistochemistry studies showed CXCL10 staining mainly in neurons throughout the brain parenchyma and only occasionally on endothelial cells, but not on astrocytes (Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008). Whereas the endothelial and glial localization of CXCL9 and CXCL10 producing cells is consistent with a role for these chemokines in the recruitment of inflammatory leucocytes to the site of parasite sequestration (intravascular infiltration without extravasation into the brain parenchyma), further research is required to determine the effect that CXCL10 upregulation might have in neuronal tissue and the effector signals responsible for this response during infection.
In addition to CXCL9 and CXCL10, the platelet-derived chemokine, platelet factor 4 (PF4 or CXCL4), which binds CXCR3B, a spliced variant of the chemokine receptor CXCR3, was found to be induced in the brain during P. berghei-ANKA infection (Srivastava et al. Reference Srivastava, Cockburn, Swaim, Thompson, Tripathi, Fletcher, Shirk, Sun, Kowalska, Fox-Talbot, Sullivan, Zavala and Morrell2008). Furthermore, PF4−/− mice showed reduced ECM mortality and T cell infiltration into the brain during infection (Srivastava et al. Reference Srivastava, Cockburn, Swaim, Thompson, Tripathi, Fletcher, Shirk, Sun, Kowalska, Fox-Talbot, Sullivan, Zavala and Morrell2008). Thus CXCL4 might contribute to the recruitment of T cells to the brain in ECM pathogenesis. In addition, CXCL4 was also found to enhance monocyte activation in response to P. berghei-ANKA infection (Srivastava et al. Reference Srivastava, Field, Aggrey, Yamakuchi and Morrell2010). The precise contribution of this response to disease severity requires further investigation.
Chemokines are not only mediators of cell trafficking during inflammation but are also involved in the recruitment of immune cells to secondary lymphoid organs whereby they enhance the development of protective responses to infection. A few studies have addressed the role that chemokines play in the control of parasite densities using rodent malaria infections. CCR2 is a receptor involved in the recruitment of various cells including activated T cells, NK cells, dendritic cells and monocytes. Whereas genetic deficiency of this chemokine does not appear to affect parasite growth during P. berghei-ANKA (Belnoue et al. Reference Belnoue, Costa, Vigario, Voza, Gonnet, Landau, Van Rooijen, Mack, Kuziel and Renia2003a ) or Plasmodium chabaudi adami infection (Weidanz et al. Reference Weidanz, LaFleur, Brown, Burns, Gramaglia and van der Heyde2010), parasite clearance was found be delayed in CCR2−/− mice infected with Plasmodium chabaudi chabaudi AS (Sponaas et al. Reference Sponaas, Freitas do Rosario, Voisine, Mastelic, Thompson, Koernig, Jarra, Renia, Mauduit, Potocnik and Langhorne2009). CCR2 is involved in the recruitment of bone marrow-derived inflammatory monocytes in response to infection (Serbina et al. Reference Serbina, Jia, Hohl and Pamer2008). The reduced control of parasitaemia observed in CCR2−/− mice correlated with significantly lower numbers of inflammatory monocytes in the spleen of these animals, suggesting these cells play a role in controlling blood-stage malaria. Monocytes as well as dendritic cells are also capable of migrating in response to CXCL12, which recognizes the CXCR4 receptor. CXCR4 blockade during Plasmodium chabaudi CR infection has also been shown to result in increased recrudescent parasitaemias (Garnica et al. Reference Garnica, Souto, Silva and de Andrade2002), however the precise mechanism by which this trafficking pathway modulates parasite density in this model requires further investigation.
CONCLUDING REMARKS
Over the past few decades, most of the work on soluble factors associated with the development of severe malaria has mainly focused on the analysis of cytokine responses both in human studies and rodent infection models. The role of chemokines in severe disease induction has only recently started to become appreciated and receive more attention. Cytokines are primarily secreted by immune cells and their main function is modulation of immune responses. Unlike cytokines, chemokines are secreted by a broad range of cellular sources in lymphoid organs and/or inflamed tissue and they appear to control a range of diverse processes including leucocyte trafficking, angiogenesis and tissue remodelling. It is then possible that the cellular sources of chemokines produced in response to malaria differ in inflamed tissue and lymphoid organs and that they control different processes. Further research is needed to fully appreciate the contribution of these chemokine-mediated processes to severe malaria and the precise mechanisms by which inflammatory chemokines exacerbate disease.
Our current knowledge on human PM supports the idea that the intravillous infiltrate that accompanies parasite sequestration mainly consists of monocytes and macrophages. Consistent with this observation, elevated levels of monocyte-attracting β-chemokines such as CCL2 and CCL3 have been found in infected placentae and their production appears to be associated with cellular infiltration (Abrams et al. Reference Abrams, Brown, Chensue, Turner, Tadesse, Lema, Molyneux, Rochford, Meshnick and Rogerson2003), strongly suggesting that chemokine secretion in the placenta during infection recruits monocytes to the intervillous space (Fig. 1).
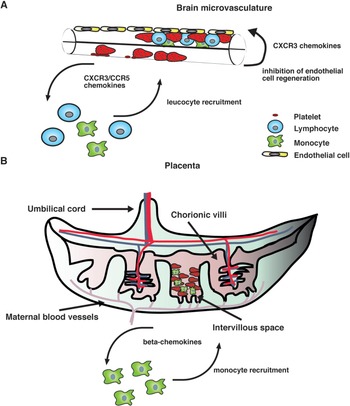
Fig. 1. A hypothetical model of action of chemokines in human severe malaria syndromes. (A) After binding to the brain microvasculature, sequestered pRBC induce activation of vascular endothelial cells, which results in the release of inflammatory cytokines as well as CXCR3 and CCR5 binding chemokines. It is possible that local production of these chemokines stimulates the accumulation of CXCR3+ and CCR5+ leucocytes. In addition, some CXCR3 chemokines such as CXCL10 that have angiostatic activity could inhibit endothelial cell regeneration of the brain microvasculature, thereby compromising the integrity of the blood–brain barrier. (B) In the placenta, both maternal and fetal cells might contribute to the production of β-chemokines in response to infection. These mediators stimulate the recruitment of monocytes and macrophages to the intravillous space, which appears to be associated with adverse pregnancy outcomes.
On the other hand, mainly CXCR3 and CCR5 binding chemokines have been found to be associated with CM severity in humans (Armah et al. Reference Armah, Wilson, Sarfo, Powell, Bond, Anderson, Adjei, Gyasi, Tettey, Wiredu, Tongren, Udhayakumar and Stiles2007). Although the precise mechanism by which these chemokines contribute to human disease remains unclear, it is possible that their local production in the brain during infection might stimulate the accumulation of CXCR3+ and CCR5+ leucocytes (Fig. 1). Consistent with this hypothesis, increased CXCR3 and CCR5 mRNA levels have been observed in brain samples from children that have succumbed to P. falciparum-mediated CM (Sarfo et al. Reference Sarfo, Singh, Lillard, Quarshie, Gyasi, Armah, Adjei, Jolly and Stiles2004). In addition to their chemotactic function, some inflammatory chemokines such as CXCL10 have been shown to exert angiostatic activity (Angiolillo et al. Reference Angiolillo, Sgadari, Taub, Liao, Farber, Maheshwari, Kleinman, Reaman and Tosato1995; Luster et al. Reference Luster, Greenberg and Leder1995; Romagnani et al. Reference Romagnani, Annunziato, Lasagni, Lazzeri, Beltrame, Francalanci, Uguccioni, Galli, Cosmi, Maurenzig, Baggiolini, Maggi, Romagnani and Serio2001). It is then possible that locally produced CXCL10 may inhibit endothelial cell regeneration of the brain microvasculature, thereby compromising the integrity of the blood–brain barrier (Fig. 1). In support of this view, the angiogenic factor VEGF has been found to be protective against CM-associated mortality (Armah et al. Reference Armah, Wilson, Sarfo, Powell, Bond, Anderson, Adjei, Gyasi, Tettey, Wiredu, Tongren, Udhayakumar and Stiles2007; Jain et al. Reference Jain, Armah, Tongren, Ned, Wilson, Crawford, Joel, Singh, Nagpal, Dash, Udhayakumar, Singh and Stiles2008).
Although rodent malaria infection models are not perfect, CXCR3 and CCR5 chemokines (such as CXCL10 and CCL4) that were identified as biomarkers of CM in humans (Armah et al. Reference Armah, Wilson, Sarfo, Powell, Bond, Anderson, Adjei, Gyasi, Tettey, Wiredu, Tongren, Udhayakumar and Stiles2007) have also been found to participate in ECM induction in mice (Campanella et al. Reference Campanella, Tager, El Khoury, Thomas, Abrazinski, Manice, Colvin and Luster2008; Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009). Thus animal models appear to provide a fast and cost-effective resource to investigate chemokine-mediated mechanisms of disease and for assessment of potential therapeutic interventions (Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009; Wilson et al. Reference Wilson, Solomon, Anderson, Patrickson, Pitts, Bond, Liu and Stiles2013). Moreover, good mouse models of SMA and PM have been developed (Evans et al. Reference Evans, Hansen, van Rooijen, Buckingham and Schofield2006; Neres et al. Reference Neres, Marinho, Goncalves, Catarino and Penha-Goncalves2008; Rodrigues-Duarte et al. Reference Rodrigues-Duarte, de Moraes, Barboza, Marinho, Franke-Fayard, Janse and Penha-Goncalves2012) and are now available to explore the involvement of chemokines in these disease syndromes.
Current views in the field propose that the same inflammatory responses that contribute to severe malaria might be also involved in the control of parasitaemia. This concept has discouraged the use of anti-cytokine treatments (van Hensbroek et al. Reference van Hensbroek, Palmer, Onyiorah, Schneider, Jaffar, Dolan, Memming, Frenkel, Enwere, Bennett, Kwiatkowski and Greenwood1996) as adjunctive therapy for complicated malaria cases as it might reduce inflammation but also result in immunosuppression, which could be detrimental for the control of infection. Anti-chemokine therapies (Nie et al. Reference Nie, Bernard, Norman, Amante, Lundie, Crabb, Heath, Engwerda, Hickey, Schofield and Hansen2009) are emerging as potential safe therapeutic alternatives to improve outcomes of severe malaria cases during treatment with anti-malarial drugs, as they alleviate organ-specific inflammation without inducing a generalized immunosuppression of the host.
FINANCIAL SUPPORT
Supported by the Australian Government National Health and Medical Research Council IRIISS and Project Grant 1031212.





 Open access
Open access