



Introduction
Mongolian gazelles (Procapra gutturosa), a species native to the temperate grasslands of Eurasia, are important herbivores. In the 1970s, there were over 3 million wild Mongolian gazelles in the grasslands of Inner Mongolia, China. However, the population has declined to approximately 1000 individuals today (Ito et al., Reference Ito, Miura, Lhagvasuren, Enkhbileg, Takatsuki, Tsunekawa and Jiang2006; Shi et al., Reference Shi, Yang, Cha, Lyu, Wang, Zhou, Dong, Dou and Zhang2023). Based on the rate and extent of population decline, Mongolian gazelles have been classified as a National First-class Protected Animal in China (Jiang et al., Reference Jiang, Wu, Liu, Jiang, Zhou, Hu and Ding2021). Mongolian gazelles are primarily distributed in the eastern grasslands of Mongolia and in Hulunbuir, Xilingol and Ulanqab in China, with a concentrated presence in the border regions between the 2 countries. They also have extensive migratory movements, typically undergoing large-scale migrations in spring and autumn. During winter, a significant number of Mongolian gazelles cross the border into China for wintering before returning to Mongolia (Bi et al., Reference Bi, Li, Sheng and Wang1997). To facilitate the free migration of Mongolian gazelles and other wildlife, China has established multiple approximately wildlife corridors on the frequently traversed border fences, considering the living habits of Mongolian gazelles and the topography and water distribution in protected areas. However, the regional movements and long-distance migrations of wildlife can significantly increase the cross-border transmission risks of infectious diseases, particularly in the context of global climate change and urbanization. The distribution areas, movement patterns and migration routes of wildlife may undergo corresponding changes, leading to accelerated disease outbreaks, especially in border regions. Furthermore, due to Mongolia being an endemic area for foot-and-mouth disease (McFadden et al., Reference McFadden, Tsedenkhuu, Bold, Purevsuren, Bold and Morris2015), and peste des petits ruminants (Benfield et al., Reference Benfield, Hill, Shatar, Shiilegdamba, Damdinjav, Fine, Willett, Kock and Bataille2021), the risk of disease transmission into China through infected Mongolian gazelles poses an increasing threat to public health security.
Zoonotic intestinal protozoan parasites primarily spread through waterborne or soil-transmitted, with human infections typically originating from a variety of domestic and wild animals. Consequently, these diseases pose significant public health challenges to both humans and wildlife, especially in poor and developing countries. (Belete et al., Reference Belete, Kassa and Baye2021; Maganga et al., Reference Maganga, Makouloutou-Nzassi, Boundenga, Maganga Landjekpo, Bangueboussa, Ndong Mebaley, Mounioko and Gbati2023). Four zoonotic protozoan intestinal parasites-Cryptosporidium spp., Cystoisospora belli, Blastocystis sp. and Cyclospora cayetanensis, are widespread threats to human health worldwide. Among these, Cryptosporidium spp. is a widespread zoonotic parasite known to cause global outbreaks of waterborne and foodborne gastroenteritis. Human infection with Cryptosporidium spp. generally does not display obvious clinical symptoms, but severe cases can lead to changes in coat colour, loss of appetite, wasting, diarrhoea and even death (Cacciò and Chalmers, Reference Cacciò and Chalmers2016; Zahedi and Ryan, Reference Zahedi and Ryan2020). One notable characteristic of Cryptosporidium is its extensive genetic variation, resulting in the existence of more than 38 species and over 60 genotypes of this parasite (Feng et al., Reference Feng, Ryan and Xiao2018). Cystoisospora belli, previously known as Isospora belli, is a parasite responsible for causing an intestinal disease called cystoisosporiasis. This protozoan parasite is opportunistic and often affects immunocompromised individuals, particularly those with HIV infection or living in institutional settings (Legua and Seas, Reference Legua and Seas2013; Rial-Crestelo et al., Reference Rial-Crestelo, Otero, Pérez-Ayala, Pinto and Pulido2022). Blastocystis sp. is an anaerobic single-celled intestinal parasite that can infect both animals and humans (Deng et al., Reference Deng, Yao, Liu, Zhou, Chai, Wang, Zhong, Deng, Ren, Fu, Yan, Yue and Peng2019). Blastocystis has been found to exist in approximately 1 to 2 billion people worldwide, making it highly prevalent (Tan, Reference Tan2008). Apart from humans, this parasite is frequently detected in various animal hosts, including non-human primates and other mammals such as ungulates, perissodactyls, proboscideans, rodents and marsupials, as well as birds, reptiles, amphibians, fish, annelids and insects (Wang et al., Reference Wang, Gong, Yang, Zhang, Zheng and Liu2018). Cyclospora sp. is an emerging pathogen responsible for global outbreaks of cyclosporiasis, posing a threat to the health of both humans and various animal species. Since its discovery, numerous studies have reported the molecular characteristics and zoonotic potential of Cyclospora in various animals (Ortega and Sanchez, Reference Ortega and Sanchez2010). On the other hand, C. cayetanensis has been confirmed as a zoonotic pathogen and is the only known genotype capable of infecting humans. Currently, documented hosts of Cyclospora include dogs, cattle, chickens, non-human primates and some rodents (Totton et al., Reference Totton, O'Connor, Naganathan, Martinez and Sargeant2021).
Transboundary migratory animals can serve as vectors for disease transmission, particularly when they come into close contact with humans or other animals. Mongolian gazelles are important wild animal resources, and therefore, it is necessary to further study the relationship between migratory Mongolian gazelles and diseases in order to develop effective conservation measures and reduce the risk of disease transmission. Therefore, this study aims to investigate the prevalence of common zoonotic intestinal protozoan parasites, including Cryptosporidium spp., C. belli, Blastocystis sp., and C. cayetanensis, in a large-scale population of Mongolian gazelles that migrated across the border and entered China from Mongolia in early December 2023. By studying the infection rate of those protozoan in Mongolian gazelles, the risk of zoonotic diseases can be assessed, and appropriate preventive measures can be implemented to reduce the occurrence and spread of outbreaks. This research holds immense importance for wildlife conservation, zoonotic disease prevention and control, as well as public health.
Methods and methods
Ethical statement
This study was conducted in strict accordance with the Guidelines for the Care and Use of Animals in Research published by the Institute of Zoology, Chinese Academy of Sciences.
Collection of stool samples
A population of Mongolian gazelles (approximately 4500–6000) migrating from the Sino-Mongolian border into China was selected as the target for monitoring in this study. From December 12th to 14th, 2023, within the activity area of the wild Mongolian gazelle population in the Hinggan League of Inner Mongolia, China (North Latitude N: 46°41′48.43″ East Longitude E: 119°58′30.60″), investigations were conducted by randomly sampling one specimen from each plot of land (each with an area of 20 m × 20 m) to ensure the representativeness of the sample and the accuracy of the conclusions drawn for the entire population. The collected fecal samples were immediately transported to the laboratory at the Institute of Zoology, Chinese Academy of Sciences in Beijing using dry ice and stored at −80°C for further experimentation.
Stool sample collection and DNA extraction
Total genomic DNA was extracted from 200 mg Stool according to the instruction of E.Z.N.A. ® Stool DNA Kit Stool Whole Genome Extraction Kit (OMEGA Biotek INCD4015-01, USA). The extracted Stool DNA was stored in a refrigerator at −20°C for further PCR assays.
PCR amplification
Nested PCR was employed to detect the small subunit ribosomal ribonucleic acid (SSU rRNA) gene of Cryptosporidium spp., C. belli, Blastocystis sp., and C. cayetanensis. The SSU rRNA gene of Cryptosporidium spp. (Xiao et al., Reference Xiao, Singh, Limor, Graczyk, Gradus and Lal2001), C. belli (Zhang, Reference Zhang2022), Blastocystis sp. (Wang et al., Reference Wang, Cuttell, Bielefeldt-Ohmann, Inpankaew, Owen and Traub2013) and C. cayetanensis (Li et al., Reference Li, Kiulia, Mwenda, Nyachieo, Taylor, Zhang and Xiao2011) were individually examined using nested PCR following established protocols. Detailed primer information and PCR procedures can be found in Table 1. Each amplification reaction included both positive and negative controls. The amplified DNA fragments underwent electrophoresis on a 1.5% agarose gel with Gel-Red. Subsequently, the resulting electrophoresis patterns were visualized, analysed using a gel imaging system, and documented through photographs.
Table 1. Forward and reverse primers used for the detection of pathogenic intestinal protozoan parasites in Procapra gutturosa

Gene sequencing
The secondary PCR positive products were sequenced using Sanger sequencing method, and the resulting sequences were spliced in both directions by Shenzhen Huada Gene Co Ltd (Beijing, China). Following the comparison of sequencing results, DNAMAN software was employed to correct splicing and assemble the sequences. The successfully sequenced sequences were then searched and aligned against related sequences with higher homology on the GenBank database hosted by the National Center for Biotechnology Information (NCBI). To further validate the alignment, the downloaded sequences and sequencing results were assembled and compared using Clustal X2.1 software.
Analysis of the evolutionary relationships
As reference genes, we selected host genotypes and environment sequences of Cryptosporidium sp., including chipmunk genotype I, muskrat genotype II, Brandt's vole, Microtus arvalis, chipmunk, tyzzeri, meleagridis, felis, andersoni, hominis, ubiquitum and other taxa. Phylogenetic tree was constructed based on SSU rRNA gene using MEGA9.0 software and the Neighbour-joining method, employing the bootstrap test with 1000 replicates and the Tamura 3-parameter model. The outgroup consisted of Eimeria sp. (GenBank: KT305927.1).
For C. belli genotypes, reference genes were selected from samples isolated from humans, Columba livia domestica, Parus major, Turdus migratorius, domestic canaries and other taxa. Phylogenetic tree was constructed based on SSU rRNA gene using MEGA9.0 software and the Neighbour-joining method, employing the bootstrap test with 1000 replicates and the Jukes–Cantor model. The outgroup consisted of Giardia intestinalis (GenBank: OR689423.1).
For Blastocystis sp. genotypes, reference genes were selected from genotypes including ST1, ST2, ST3, ST4, ST5, ST6, ST7, ST8, ST9, ST10, ST12, ST13 and ST14 serotypes, isolated from Homo sapiens, Cervus nippon, Alouatta palliata aequatoriali, Camelus dromedarius, Macaca fuscata, Moschus berezovskii, Anser cygnoides, Wallabia bicolor mastersii, Gallus domesticus, Ruttus novercious, Rusa unicolor, Elaphodus cephalopus and Ovis aris. Phylogenetic tree was constructed based on SSU rRNA gene using MEGA9.0 software and the Neighbour-joining method, employing the bootstrap test with 1000 replicates and the Jukes–Cantor model. The outgroup consisted of Histomonas meleagridis (GenBank: AJ920323.1).
For C. cayetanensis genotypes, reference genes were selected from samples isolated from H. sapiens, Macaca mulatta, Du-long cattle, dairy cattle farm, Holstein cattle, dairy cattle, Macaca fascicularis, baboon and other taxa. Phylogenetic tree was constructed based on SSU rRNA gene using MEGA9.0 software and the Neighbour-joining method, employing the bootstrap test with 1000 replicates and the Tamura 3-parameter model. The outgroup consisted of Entamoeba histolytica (GenBank: OR236732.1).
Results
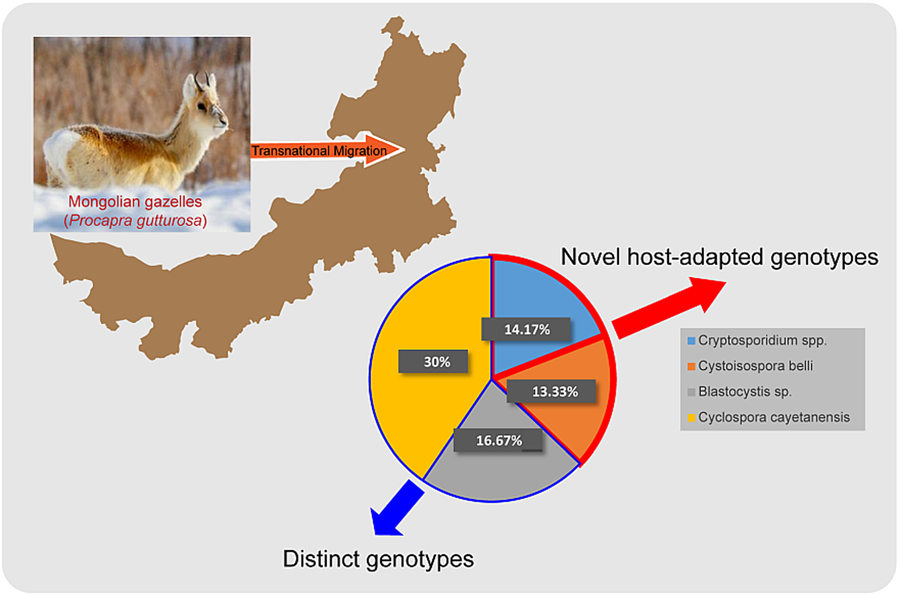
The infection rates of Cryptosporidium spp., C. belli, Blastocystis sp., and C. cayetanensis in Mongolian gazelles
Positive samples were determined based on successful sequence alignment in the sequencing results. Among the 120 samples, the infection rates results indicated that Cryptosporidium spp. was detected in 14.17% (17/120), followed by C. belli in 13.33% (16/120), Blastocystis sp. in 16.67% (20/120) and C. cayetanensis in 30.00% (36/120) (Table 2). All PCR-positive samples from the Cryptosporidium SSU rRNA gene were sequenced, and 2 complete and representative sequences were submitted to NCBI GenBank (Accession Numbers: PP159013 and PP159016) after excluding poor quality sequences due to sequence fragmentation, incompleteness, or inaccurate sequence content. Similarly, the sequences of the SSU rRNA gene locus of C. belli were submitted to GenBank, acquiring the sequence accession numbers: PP159019 and PP159020. Additionally, the sequences of the SSU rRNA gene locus of Blastocystis sp. were sequenced and submitted to NCBI GenBank, resulting in the assignment of sequence accession numbers: PP159017 and PP159018. Lastly, the sequences of the SSU rRNA gene locus of C. cayetanensis were sequenced and submitted to NCBI GenBank, acquiring the sequence accession numbers: PP159021 and PP159025.
Table 2. The prevalence (No. Positive/No. Tested) of pathogenic intestinal protozoa in Procapra gutturosa

Evolutionary relationships analysis of SSU rRNA gene in Cryptosporidium spp. isolated from Mongolian gazelles
A phylogenetic tree (Fig. 1) was constructed using the SSU rRNA gene sequence of a reference Cryptosporidium spp. The Cryptosporidium spp. genotypes discovered in Mongolian gazelles (PP159013 and PP159016) formed a distinct branch (97 to 98% homology), indicating their potential as host-specific genotypes for Mongolian gazelles. Based on the molecular characteristics and host specificity differences, we designated the newly identified genotype as Cryptosporidium Mongolian gazelle genotype.

Figure 1. The Evolutionary relationships of Cryptosporidium sp. genotypes identified in the present study and other known genotypes and species on GenBank were inferred by a Neighbour-joining method of SSU rRNA gene using the bootstrap test (1000 replicates) and Tamura 3-parameter model method. The Eimeria sp. (GenBank: KT305927.1) were used as the outgroup. The Solid red lines and red 5-pointed stars represent novel genotypes identified in this study.
Evolutionary relationships analysis of SSU rRNA gene in C. belli isolated from Mongolian gazelles
A phylogenetic tree (Fig. 2) was constructed using the SSU rRNA gene sequence of a reference C. belli. The C. belli genotypes found in Mongolian gazelles (PP159019 and PP159020), which formed a distinct branch, had a homology of 99%, suggesting their potential as host-specific genotypes for Mongolian gazelles. Considering the molecular characteristics and differences in host specificity, we designated the newly identified genotype as C. belli Mongolian gazelle genotype.

Figure 2. The Evolutionary relationships of Cystoisospora belli genotypes identified in the present study and other known genotypes and species on GenBank were inferred by a Neighbour-joining method of SSU rRNA gene using the bootstrap test (1000 replicates) and Jukes–Cantor model method. The Giardia intestinalis (GenBank: OR689423.1) were used as the outgroup. The Solid red lines and red 5-pointed stars represent novel genotypes identified in this study.
Evolutionary relationships analysis of SSU rRNA gene in Blastocystis sp. isolated from Mongolian gazelles
A phylogenetic tree (Fig. 3) was constructed by comparing the SSU rRNA gene sequence of Blastocystis sp. with the reference sequence. Based on the clustering results, the genotype of Blastocystis sp. (PP159017) was most closely related (88% to 89% homology) to Rusa unicolor (AB070997.1) ST2 serotype, clustered together with it, leading us to designate this newly discovered genotype as serotype ST2. Conversely, the genotype of Blastocystis sp. (PP159018) was most closely related (79% to 80% homology) with the Japanese macaque (MT114848.1) and E. cephalopus (MW410724.1) ST13 serotypes, and clustered with them, prompting us to assign this newly discovered genotype as serotype ST13.

Figure 3. The Evolutionary relationships of Blastocystis sp. genotypes identified in the present study and other known genotypes and species on GenBank were inferred by a Neighbour-joining method of SSU rRNA gene using the bootstrap test (1000 replicates) and Jukes–Cantor model method. The Histomonas meleagridis (GenBank: AJ920323.1) were used as the outgroup. The Solid red lines and red 5-pointed stars represent novel genotypes identified in this study.
Evolutionary relationships analysis of SSU rRNA gene in C. cayetanensis isolated from Mongolian gazelles
A phylogenetic tree (Fig. 4) was constructed by comparing the SSU rRNA gene sequence of C. cayetanensis with the reference sequence. Based on the clustering results, the genotype of C. cayetanensis isolates from Mongolian (PP159021 and PP159025) were most closely related (94 to 95% homology) to primates (AF061566.1), with which they clustered.

Figure 4. The Evolutionary relationships of Cyclospora cayetanensis genotypes identified in the present study and other known genotypes and species on GenBank were inferred by a Neighbour-joining method of SSU rRNA gene using the bootstrap test (1000 replicates) and Tamura 3-parameter model method. The Entamoeba histolytica (GenBank: OR236732.1) were used as the outgroup. The Solid red lines and red 5-pointed stars represent novel genotypes identified in this study.
Discussion
Transboundary migratory animals play a crucial role in disease outbreaks by facilitating the transmission and spread of pathogens during their long-distance migrations. They can potentially serve as reservoirs for various novel pathogens, introducing them to new geographic areas as they traverse different regions (Uhlemann et al., Reference Uhlemann, McAdam, Sullivan, Knox, Khiabanian, Rabadan, Davies, Fitzgerald and Lowy2017). In the case of Mongolian gazelles, many new pathogens have been discovered. For instance, an undescribed chewing louse of the genus Damalinia was collected from Mongolian gazelles (Lux et al., Reference Lux, Mix and Zedev1997). Furthermore, in 2014, Taenia spp. and Anaplasma were reported for the first time in Mongolian gazelles (Li et al., Reference Li, Chen, Liu, Liu, Yang, Li, Li, Ren, Niu, Guan, Luo and Yin2014). Research conducted on the nematode composition in the digestive tract of Mongolian gazelles revealed the presence of Orloffia bisonis, Marshallagia mongolica, Nematodirus archari, Nematodirus andreevi, Trichostrongylus colubriformis and Trichostrongylus probolurus. Notably, N. archari and N. andreevi were found for the first time in Mongolian gazelles, and all gastrointestinal nematode species found in Mongolian gazelles have been recorded in domestic animals within Mongolia (Kuznetsov et al., Reference Kuznetsov, Khrustalev, Batchimeg and Danzan2014). During migration, animals undergo physical decline, increased energy consumption, and heightened immune system stress, making them more susceptible to disease. Moreover, in comparison to other animal species, Mongolian gazelles may have more frequent close contact and interactions, potentially resulting in an increased transmission of parasites and other infectious diseases (Kuznetsov et al., Reference Kuznetsov, Khrustalev, Batchimeg and Danzan2014). In 1990, over 1000 Mongolian gazelles migrating from Mongolia to grazing areas in China died due to infection with Pasteurellosis (Yuan et al., Reference Yuan, Ha and Lou1991). Research conducted testing for foot-and-mouth disease virus in serum samples collected from Mongolian gazelles in eastern Mongolia and found that these viruses in Mongolian gazelles were likely due to spill-over from livestock (Nyamsuren et al., Reference Nyamsuren, Joly, Enkhtuvshin, Odonkhuu, Olson, Draisma and Karesh2006). Studying the spillover of diseases in migratory cross-border Mongolian gazelles is of significant importance for understanding disease transmission and pathogenesis (Wang et al., Reference Wang, Qin, Bao, Qiao, Yang and Zhao2009). Additionally, in the central-Mongolian border region of Inner Mongolia, there is a large population of rodents which also serve as natural hosts for parasite transmission (Feng et al., Reference Feng, Chang, Wang, Huang, Han and He2020), these rodents may have direct or indirect contact with Mongolian gazelles in geographically overlapping areas, thereby increasing the risk of disease spillover. However, there is a lack of sufficient research on the impact of cross-border migratory terrestrial animals on disease outbreaks, particularly in the case of Mongolian gazelles, when compared to migratory birds that can carry avian influenza viruses (van der Kolk, Reference van der Kolk2019). This is the reason why we did not conduct a comparative analysis and discussion of infection rates between hosts.
Most morphological differences among protozoan parasites cannot be distinguished by the naked eye, and relying solely on oocyst morphology identification has limitations. To overcome this deficiency, genetic analysis using molecular identification methods can be employed to analyse genetic variations among different species or within species of protozoan parasites (Xiao, Reference Xiao2010; Ryan et al., Reference Ryan, Fayer and Xiao2014). The 18S rRNA gene is responsible for encoding the multicopy repeat sequence of the SSU rRNA, which is approximately 1850 bp in length. It serves as a marker gene with informative and functional roles, making it the most widely used genotyping tool for identifying host-adapted genotypes of protozoan parasites (Xiao and Feng, Reference Xiao and Feng2017). In this study, we discovered new host-adapted genotypes of Cryptosporidium spp. and C. belli in Mongolian gazelles. Host adaptation is a common phenomenon in the genus Cryptosporidium, and specific genotypes are often associated with specific animal groups (Xiao et al., Reference Xiao, Sulaiman, Ryan, Zhou, Atwill, Tischler, Zhang, Fayer and Lal2002). Cryptosporidium has been found to have various host-adapted genotypes in many studies, especially among wildlife (Hu et al., Reference Hu, Wang, Zhang, Wang, Xing, Han and He2022). Studies on C. belli are currently mainly focused on humans, with fewer studies conducted on wildlife (Xu et al., Reference Xu, Jiang, Liu, Jiang, Wang, Zhou, Shen and Cao2021). The emergence of host-adapted genotypes is the result of long-term coevolutionary interactions between hosts and parasites (Vorburger and Perlman, Reference Vorburger and Perlman2018). This not only helps to shape the host immune system but also contributes to parasite genetic heterogeneity (Møller, Reference Møller2008; Schwensow et al., Reference Schwensow, Axtner and Sommer2011; Soler, Reference Soler2014). Specific environmental conditions determine how any genotype of one species affects the adaptations of another species (Laine, Reference Laine2009). Host-adapted genotypes are more likely to be found in the presence of co-evolving parasites if adaptation has a significant benefit to the host but little or no detrimental effect on the parasite (Ebert, Reference Ebert2005). Therefore, elucidating the occurrence of novel host-adapted genotypes in wildlife is of great significance for understanding host–parasite relationships, disease transmission and host evolution.
Furthermore, to date, a total of 32 genotypes (STs) of Blastocystis sp. have been reported based on the polymorphism within the small subunit ribosomal RNA (SSU rRNA) gene (Liu et al., Reference Liu, Ni, Wang, Li, Ge, Yang, Qi and Zhang2022). Additionally, we identified 2 new genotypes of Blastocystis sp., 1 belonging to ST2 and the other belonging to ST13. Previous studies have shown that Blastocystis sp. ST2 is commonly found in primates, while ST13 has only been detected in wildlife so far (Cian et al., Reference Cian, El Safadi, Osman, Moriniere, Gantois, Benamrouz-Vanneste, Delgado-Viscogliosi, Guyot, Li, Monchy, Noël, Poirier, Nourrisson, Wawrzyniak, Delbac, Bosc, Chabé, Petit, Certad and Viscogliosi2017). This indicates the presence of multiple genotypes of Blastocystis sp. in Mongolian gazelles, posing a risk of zoonotic infection. In addition, a total of 19 genotypes of Cyclospora have been reported, including those capable of infecting non-human primates (Li et al., Reference Li, Ye, Arrowood, Ma, Wang, Xu, Feng and Xiao2015). Among them, C. cayetanensis has been confirmed as a zoonotic pathogen and is the only known genotype capable of infecting humans (Ortega and Sanchez, Reference Ortega and Sanchez2010). The clustering of the C. cayetanensis genotype we identified with sequences obtained from non-human primate hosts in the same branch suggests a close genetic evolutionary relationship. This suggests that Mongolian gazelles may have a connection to their contact with the human environment.
In summary, this study represents the first time to investigate the infection status of 4 zoonotic intestinal protozoan parasites (Cryptosporidium spp., C. belli, Blastocystis sp. and C. cayetanensis) in migratory Mongolian gazelles. Among these, infection with C. cayetanensis is higher and was the dominant species in this study. The identification of recently discovered host-adapted genotypes within this study suggests that the Mongolian gazelle population may harbour a multitude of uncharacterized or novel pathogen genotypes, increasing the likelihood of pathogen spillover. However, investigations into virulence are limited by the scarcity of parasites isolated from samples, which will be a primary focus of our subsequent research. Therefore, it is crucial to enhance active surveillance measures and conduct comprehensive investigations into reverse pathogenesis on unique wildlife species in cross-border regions.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Authors’ contributions
H.H, S.G. and B.H. contributed to the conception of the study; B.H., Y.C, L.H., D.X. and S.G. contributed significantly to collect samples and perform the experiment; G.L., X.A. and Y.X. contributed significantly to analysis and manuscript preparation; S.H. and W.L. helped perform the analysis with constructive discussions; S.G. performed the data analyses and wrote the manuscript.
Financial support
This study was supported by the National Key Research and Development Program of China (No. 2022YFC2601602), the Major Program of National Natural Science Foundation of China (No. 32090023), and National Forestry and Grassland Administration, China.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical standards
The study and collection of samples were approved by the Animal Ethics Committee of the Institute of Zoology, Chinese Academy of Sciences (approval number: IOZ-15042).







 Open access
Open access