



Introduction
Trypanosomes are members of the Euglenozoa, a group of organisms within the Incertae sedis Eukarya (ex-Excavata) supergroup (Adl et al., Reference Adl, Bass, Lane, Lukeš, Schoch, Smirnov, Agatha, Berney, Brown, Burki, Cárdenas, Čepička, Chistyakova, Campo, Dunthorn, Edvardsen, Eglit, Guillou, Hampl, Heiss, Hoppenrath, James, Karnkowska, Karpov, Kim, Kolisko, Kudryavtsev, Lahr, Lara, Le Gall, Lynn, Mann, Massana, Mitchell, Morrow, Park, Pawlowski, Powell, Richter, Rueckert, Shadwick, Shimano, Spiegel, Torruella, Youssef, Zlatogursky and Zhang2019). These organisms are likely to have branched very early during evolution, which may explain the vast number of unorthodox features that define their biology (Navarro et al., Reference Navarro, Peñate and Landeira2007; Adl et al., Reference Adl, Bass, Lane, Lukeš, Schoch, Smirnov, Agatha, Berney, Brown, Burki, Cárdenas, Čepička, Chistyakova, Campo, Dunthorn, Edvardsen, Eglit, Guillou, Hampl, Heiss, Hoppenrath, James, Karnkowska, Karpov, Kim, Kolisko, Kudryavtsev, Lahr, Lara, Le Gall, Lynn, Mann, Massana, Mitchell, Morrow, Park, Pawlowski, Powell, Richter, Rueckert, Shadwick, Shimano, Spiegel, Torruella, Youssef, Zlatogursky and Zhang2019). Euglenozoa include Euglenids, Phytomonads and Trypanosomatids, free-living phagotrophs, plant and animal parasites, respectively (Adl et al., Reference Adl, Bass, Lane, Lukeš, Schoch, Smirnov, Agatha, Berney, Brown, Burki, Cárdenas, Čepička, Chistyakova, Campo, Dunthorn, Edvardsen, Eglit, Guillou, Hampl, Heiss, Hoppenrath, James, Karnkowska, Karpov, Kim, Kolisko, Kudryavtsev, Lahr, Lara, Le Gall, Lynn, Mann, Massana, Mitchell, Morrow, Park, Pawlowski, Powell, Richter, Rueckert, Shadwick, Shimano, Spiegel, Torruella, Youssef, Zlatogursky and Zhang2019). Trypanosomatids include several parasitic protozoa that cause a huge health and economic burden amongst the world's poorest populations; these include Leishmania sp, Trypanosoma cruzi, Trypanosoma brucei, Trypanosoma congolense and Trypanosoma vivax. Notably, climate change, increased mobility and mass migration pose great challenges to our ability to control diseases caused by these organisms, rendering the need for new drugs to fight new parasite strains and resistance emergence imperative. Therefore, a detailed molecular understanding of fundamental aspects of their cell biology, gene expression, metabolism and interaction with the hosts is critical to design effective control strategies.
Trypanosoma brucei is the causative agent of sleeping sickness and nagana in humans and cattle, respectively, and has been used for decades as a model organism for this group mostly given its genetic tractability and available tools for reverse and forward genetics (Djikeng et al., Reference Djikeng, Shi, Tschudi and Ullu2001; Alsford et al., Reference Alsford, Turner, Obado, Sanchez-Flores, Glover, Berriman, Hertz-Fowler and Horn2011; Dean et al., Reference Dean, Sunter and Wheeler2017; Rico et al., Reference Rico, Jeacock, Kovářová and Horn2018).
Trypanosoma brucei is transmitted through the bite of a tsetse fly and rapidly differentiates into ‘slender’ bloodstream forms (BSFs) in the mammalian host. The slender forms are capable of sensing the population density, which triggers differentiation into stumpy forms. The latter are pre-adapted to life in the tsetse, where they will eventually differentiate into the procyclic forms. In the mammalian host, besides the BSFs, these parasites can occupy multiple tissues (brain, adipose tissue, skin, etc.), some recently identified as important reservoirs (Capewell et al., Reference Capewell, Cren-Travaillé, Marchesi, Johnston, Clucas, Benson, Gorman, Calvo-Alvarez, Crouzols, Jouvion, Jamonneau, Weir, Stevenson, O'Neill, Cooper, Swar, Bucheton, Ngoyi, Garside, Rotureau and MacLeod2016; Trindade et al., Reference Trindade, Rijo-Ferreira, Carvalho, Pinto-Neves, Guegan, Aresta-Branco, Bento, Young, Pinto, Van Den Abbeele, Ribeiro, Dias, Smith and Figueiredo2016).
Mammalian-infective T. brucei undergoes antigenic variation to successfully evade the host adaptive immune responses (Fig. 1A), similarly to other pathogens such as malaria and giardiasis causing parasites (Duraisingh and Horn, Reference Duraisingh and Horn2016). For that purpose, it relies on a vast genetic repertoire of genes that encode for their variant surface glycoprotein (>2500 VSG genes and pseudogenes), approximately one-third of its genome (Berriman et al., Reference Berriman2005; Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). There are two key features for successful antigenic variation: (1) the ability to express a single antigen from myriad possibilities (monogenic expression); (2) the ability to switch from one antigen isoform to another (Duraisingh and Horn, Reference Duraisingh and Horn2016). However, despite the vast genetic repertoire, a VSG gene can only be expressed from a limited subset of sub-telomeric transcription units known as expression-sites (ESs) (Navarro and Cross, Reference Navarro and Cross1996; Hertz-Fowler et al., Reference Hertz-Fowler, Figueiredo, Quail, Becker, Jackson, Bason, Brooks, Churcher, Fahkro, Goodhead, Heath, Kartvelishvili, Mungall, Harris, Hauser, Sanders, Saunders, Seeger, Sharp, Taylor, Walker, White, Young, Cross, Rudenko, Barry, Louis and Berriman2008; Fig. 1B). VSG-ESs are polycistronic transcription units (PTUs) that share the same DNA elements, and yet, one is active whereas the remaining are silent – a classic epigenetic paradigm (Duraisingh and Horn, Reference Duraisingh and Horn2016).
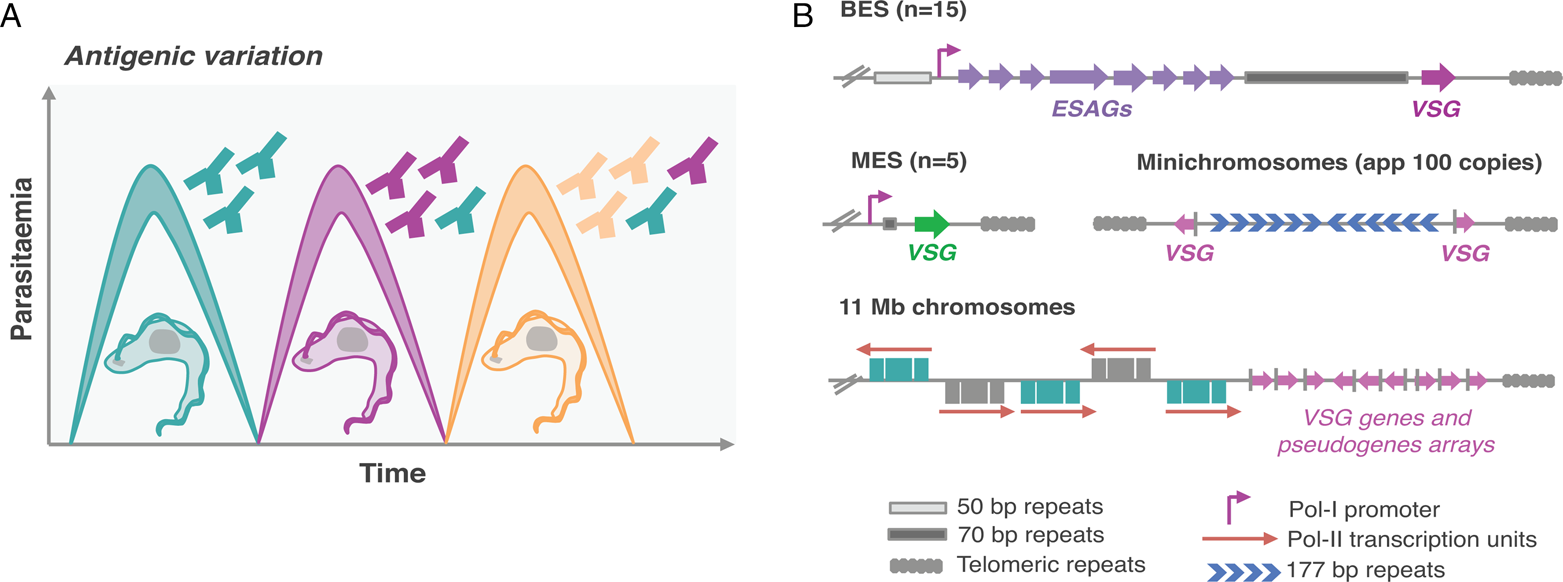
Fig. 1. Antigenic variation in T. brucei bloodstream forms. (A) Antigenic variation. Waves of parasitaemia are a hallmark of infections by African trypanosomes in mammals. This is due to waves of parasites expressing different VSG coats (different colours). VSGs are highly immunogenic, typically triggering an effective and lasting immune response (immunosuppression can occur later during infection). This illustration is a simplified depiction of the in vivo dynamics, indeed, at any time point the populations can be much more complex than represented: these may include large numbers of different clonal VSG variants. (B) Genomic organization of VSG genes. Bloodstream VSG expression-sites (BESs) contain expression-site-associated genes (ESAGs), which are located between the promoter and the 70 bp repeats. The VSG genes are near telomeric repeats. Large extensions of 50 bp repeats are located upstream of all BESs. Metacyclic VSG expression-sites (MESs) lack ESAGs and are expressed in metacyclic trypomastigotes in the salivary glands of the tsetse fly. Pol-II transcribed genes are organized in long polycistronic transcription units in the 11 megabase (Mb) size chromosomes. The arrows indicate the direction of Pol-II transcription. VSG genes or pseudogenes are organized in sub-telomeric regions of megabase chromosomes or at the telomeres of minichromosomes.
The molecular understanding of the mechanisms underpinning antigenic variation is critical as it sustains persistent infections and has greatly challenged vaccine development against these organisms. This review will be focused on nuclear compartmentalization and how it affects or might affect both antigen and global gene expression in the African trypanosome. Overall, T. brucei nuclear architecture and mechanisms for gene expression control follow some of the classic conventions but also present phenomenal dissimilarities when compared to so-called model eukaryotes.
Genome organization
Eukaryotic genomes are condensed by several orders of magnitude; such compaction is critical to fit into the nucleus of a cell. This is achieved by coiling the DNA around histones forming chromatin fibres, which are subsequently arranged into more complex high-order structures such as loops, domains and compartments (Gibcus and Dekker, Reference Gibcus and Dekker2013; Finn and Misteli, Reference Finn and Misteli2019). Several of these architectural features are conserved across the evolutionary tree, suggesting an elementary role of spatial organization in genome function and gene expression control (Foster and Bridger, Reference Foster and Bridger2005). Indeed, DNA spatial organization and compartmentalization has been found to play a key role in the regulation of gene expression and recombination in multiple organisms. In mammals on a larger scale, two major sub-nuclear compartments can be defined, one is transcription-permissive (compartment A) and the other transcription-repressive (compartment B), roughly corresponding to euchromatin and heterochromatin, respectively (Gibcus and Dekker, Reference Gibcus and Dekker2013; Finn and Misteli, Reference Finn and Misteli2019). Further, within chromatin domains known as topologically associating domains (TADs), chromatin loops modulate interactions between promoters and distal regulatory elements, ultimately impacting gene expression (Rao et al., Reference Rao, Huntley, Durand, Stamenova, Bochkov, Robinson, Sanborn, Machol, Omer, Lander and Aiden2014; Schoenfelder and Fraser, Reference Schoenfelder and Fraser2019). TADs are usually defined by boundary elements containing architectural chromatin proteins; these include cohesin, CCCTC-binding factor (CTCF) and histone variants (Millau and Gaudreau, Reference Millau and Gaudreau2011; Merkenschlager and Odom, Reference Merkenschlager and Odom2013).
The core genome of the African trypanosome
Trypanosoma brucei has a diploid genome, the haploid nuclear genome (32 Mbp) is divided into three classes of linear chromosomes: 11 pairs of megabase chromosomes (at least 1 Mbp), one to five intermediate-sized chromosomes and more than 100 minichromosomes (50–150 kbp) (Wickstead et al., Reference Wickstead, Ersfeld and Gull2004; Berriman et al., Reference Berriman2005). The megabase chromosomes contain all RNA-Polymerase-II (Pol-II) transcribed genes and VSG-ESs (Berriman et al., Reference Berriman2005; Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). The minichromosomes are highly repetitive and many also contain VSG genes (Wickstead et al., Reference Wickstead, Ersfeld and Gull2004; Fig. 1B). Additionally, T. brucei has an unknown number of highly repetitive circular extra-chromosomal DNAs of unknown function (Alsford et al., Reference Alsford, Navarro, Jamnadass, Dunbar, Ackroyd, Murphy, Gull and Ersfeld2003).
In trypanosomes, electron-dense chromatin regions can be found close to the nuclear periphery and their arrangement is developmentally regulated (Belli, Reference Belli2000; Elias et al., Reference Elias, Marques-Porto, Freymüller and Schenkman2001; Navarro et al., Reference Navarro, Peñate and Landeira2007). Indeed, in T. brucei, chromosome conformational capture (Hi-C) revealed that the transcribed chromosome core regions and the sub-telomeric regions coding for the large reservoir of silent VSG genes appear to fold into structurally distinct compartments (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018), similar to active A and silent B compartments described in mammalian cells (Schoenfelder and Fraser, Reference Schoenfelder and Fraser2019). Further, the relative interaction frequency was substantially higher across sub-telomeric regions compared to core regions, indicating that sub-telomeres are more compact than the core region. Additionally, centromeres and junctions between the core and sub-telomeres were found to be the most prominent boundaries of DNA compartments (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018).
Regarding architectural chromatin proteins, while CTCF appears to be absent in non-metazoans (Heger et al., Reference Heger, Marin, Bartkuhn, Schierenberg and Wiehe2012), the major subunit of cohesin is present in T. brucei and its depletion is lethal (Landeira et al., Reference Landeira, Bart, Van Tyne and Navarro2009). Moreover, histone variants (H3V and H4V) also function as architectural proteins in this organism (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). Indeed, studies in T. brucei H3V and H4V knockout cell lines revealed changes in global genome architecture and local chromatin configuration, which triggered switches in VSG expression (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018).
The telomeres and sub-telomeres
Genome sequences of T. brucei and Plasmodium revealed that Pol-II transcribed genes are located in the central core and antigen genes are located in sub-telomeric regions (Berriman et al., Reference Berriman2005; Otto et al., Reference Otto, Böhme, Sanders, Reid, Bruske, Duffy, Bull, Pearson, Abdi, Dimonte, Stewart, Campino, Kekre, Hamilton, Claessens, Volkman, Ndiaye, Amambua-Ngwa, Diakite, Fairhurst, Conway, Franck, Newbold and Berriman2018; Fig. 1B).
The telomere is a special functional complex at the end of linear chromosomes, consisting of tandem repeat DNA sequences and associated proteins, which can form a specialized heterochromatic structure that suppresses the expression of genes located at the sub-telomere, known as telomere position effect or telomeric silencing (Ottaviani et al., Reference Ottaviani, Gilson and Magdinier2008). Telomeres are essential for genome integrity and chromosome stability in eukaryotes and their synthesis is mainly achieved by the cellular reverse transcriptase telomerase, an RNA-dependent DNA polymerase that adds telomeric DNA to telomeres (Cong et al., Reference Cong, Wright and Shay2002). Telomerase activity was found to be absent in most normal human somatic cells, which is intimately related with the ageing process, but present in over 90% of cancerous cells (Cong et al., Reference Cong, Wright and Shay2002). Notably, telomere-binding proteins play critical roles on the maintenance of telomere length, telomere heterochromatin formation, regulation of the telomeric transcript levels, among others (Ottaviani et al., Reference Ottaviani, Gilson and Magdinier2008). The mammalian telomere complex has been well characterized and contains six core proteins that include TRF1, TRF2, TIN2, RAP1, TPP1 and POT1 (de Lange, Reference de Lange2005). Additionally, an integral component of telomeric heterochromatin is the telomeric repeat-containing RNA (TERRA), a large non-coding RNA whose transcription occurs at most or all chromosome ends. Further, R-Loops have been identified at the telomeres, these are three-stranded nucleic acid structures that contain a DNA:RNA hybrid. R-Loops can play an important role in a number of cellular functions but they can also be an instability factor (Tan and Lan, Reference Tan and Lan2020).
In the insect-stage, trypanosome telomeres tend to be close to the nuclear periphery, but this is much less pronounced in the mammalian-stage (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012). In T. brucei, besides the telomerase components (Dreesen et al., Reference Dreesen, Li and Cross2005; Sandhu et al., Reference Sandhu, Sanford, Basu, Park, Pandya, Li and Chakrabarti2013), which are critical for telomere maintenance, several other telomere proteins have been identified. Among these, TbTRF, a functional homologue of mammalian TRF2, a TbTRF-interacting factor, TIF2, RAP1 and TelAP1 (Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009; Jehi et al., Reference Jehi, Li, Sandhu, Ye, Benmerzouga, Zhang, Zhao and Li2014a, Reference Jehi, Wu and Li2014b; Reis et al., Reference Reis, Schwebs, Dietz, Janzen and Butter2018). Except for TelAP1, all the other factors are essential for cell viability; TbTRF and TbTIF2 are critical for telomere integrity and their depletion leads to an increase in double-strand breaks and increased VSG switching (Jehi et al., Reference Jehi, Li, Sandhu, Ye, Benmerzouga, Zhang, Zhao and Li2014a, Reference Jehi, Wu and Li2014b). TbRAP1 interacts with TbTRF and its depletion leads to derepression of silent VSG-ESs in the mammalian-infective stage, but also in insect-stage cells, where VSG expression is developmentally shut down (Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009). Further, TbRAP1-mediated silencing has a stronger impact on telomere proximal genes (Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009). Moreover, by associating with telomere chromatin, TbRAP1 also suppresses the expression of the TERRA transcripts and telomeric R-Loops, consistent with a role on telomere integrity (Nanavaty et al., Reference Nanavaty, Sandhu, Jehi, Pandya and Li2017). Recent studies on T. brucei ribonuclease H enzymes, endonuclease enzymes that catalyse the cleavage of RNA in an RNA/DNA substrate, also showed that R-loops at the telomere and the sub-telomere affect VSG switching frequencies (Briggs et al., Reference Briggs, Crouch, Lemgruber, Lapsley and McCulloch2018, Reference Briggs, Crouch, Lemgruber, Hamilton, Lapsley and McCulloch2019).
Interestingly, the nuclear phosphatidylinositol 5-phosphatase (PIP5Pase), part of the inositol phosphate pathway, has been recently shown to interact with TbRAP1 in a ~0.9-MDa complex (Cestari et al., Reference Cestari, McLeland-Wieser and Stuart2019). The inositol phosphate pathway regulates several cellular processes in eukaryotes including chromatin remodelling and gene expression, and had been shown to have a role on telomere silencing and VSG monogenic expression in T. brucei (Cestari and Stuart, Reference Cestari and Stuart2015).
In summary, in T. brucei (similarly to Plasmodium), Pol-II transcribed genes are located in the central core whereas the antigen genes are located in sub-telomeric regions (Berriman et al., Reference Berriman2005; Otto et al., Reference Otto, Böhme, Sanders, Reid, Bruske, Duffy, Bull, Pearson, Abdi, Dimonte, Stewart, Campino, Kekre, Hamilton, Claessens, Volkman, Ndiaye, Amambua-Ngwa, Diakite, Fairhurst, Conway, Franck, Newbold and Berriman2018). This chromosome partitioning may be important to fine-tune recombination in regions that encode for antigens and to ensure that all but one antigen is repressed. Similarly to Plasmodium, there is a large amount of evidence that supports a role for telomeric chromatin in VSG gene silencing (Duraisingh and Horn, Reference Duraisingh and Horn2016). Moreover, the sub-telomeric location of VSG-ESs is thought to favour recombination, since these sites are rather unstable (Glover et al., Reference Glover, Alsford and Horn2013). Recombination-based and transcriptional mechanisms can lead to VSG switching, but undoubtedly recombination makes the largest contribution in T. brucei compared to Plasmodium. Indeed, telomere integrity and stability impacts VSG switching frequencies and has been also proven critical to maintain VSG monogenic expression (reviewed by Saha et al., Reference Saha, Nanavaty and Li2020). Notably, one of the many remaining outstanding questions is how the active VSG-ES escapes telomeric silencing.
Remarkably, the active VSG-ES and the silent VSG-ESs reside within distinct nuclear compartments; the importance of nuclear compartmentalization on global gene expression control and VSG expression, in particular, will be addressed in the next chapter.
Nuclear compartmentalization
The nucleus is a double lipid bilayer enclosed organelle, which separates genomic DNA from the rest of the cell. Its architecture shields the genome from the sources of damage whilst providing opportunities for gene expression regulation (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019). There is ample evidence in multiple eukaryotes that the transcriptional activity of genes is influenced by nuclear organization, which changes during differentiation and development. Indeed, the regulated expression of genes during development is influenced by the availability of regulatory proteins and the accessibility of the DNA to the transcriptional machinery (Finn and Misteli, Reference Finn and Misteli2019). In eukaryotes, heterochromatin, which is highly compact, is mainly located at the nuclear periphery, whereas the less compact euchromatin occupies a more interior nuclear position.
Additionally, key nuclear functions such as transcription, replication or RNA processing are not homogeneously distributed throughout the nucleus and can be compartmentalized. Such compartmentalization within the nucleoplasm enables functional specialization, separation of conflicting processes as well as increasing the concentration of specific factors at their target point of action (Finn and Misteli, Reference Finn and Misteli2019).
Two main models of nuclear organization emerged in the past. A deterministic model proposed that specific structural elements in the nucleus assembled into a scaffold that was then used by transcriptional processes, resulting in transcriptional compartmentalization, which was independent of active processes. Chromosome position would therefore be maintained by interactions with the scaffold (Misteli, Reference Misteli2007). In striking contrast, in a self-organization model, functional sites were formed depending on the gene activation status and without the need for predefined structures; chromosome position would therefore be established by chromatin itself and interactions with functional sites. Arguably, experimental data from many model systems strongly favour self-organization models over deterministic models. For instance, perturbing nuclear lamins, one of the prime structural components of the nucleus, has a modest impact on the spatial organization of transcription and pre-mRNA splicing sites, arguing against deterministic models (Spann et al., Reference Spann, Moir, Goldman, Stick and Goldman1997). Conversely, perturbation of most active nuclear processes results in rapid chromatin architectural changes, consistent with self-organization models (Misteli, Reference Misteli2007).
The nuclear periphery
At the nuclear periphery, there is a meshwork, designated nuclear lamina (NL), which in mammals is composed mainly by nuclear lamins. A growing number of nuclear proteins are known to bind lamins and are implicated in nuclear and chromatin organization, mechanical and genome stability, cell signalling, gene regulation, among others (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008). Notably, many molecules must be able to traffic between the nucleus and the cytoplasm, rendering nucleo-cytoplasmic transport absolutely critical for cell survival. The trafficking of macromolecules in and out of the nucleus occurs through nuclear pore complexes (NPCs) (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019).
Nuclear pore
NPCs are massive macromolecular assemblies! In humans, each NPC consists of ~1000 protein subunits, designated nucleoporins, rendering it one of the largest protein complexes in nature (~110 MDa). Each NPC is located in and stabilizes an ~800 Å-wide nuclear pore, which is generated by the fusion between the inner and outer nuclear membranes (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019).
NPCs are critical to maintain the nuclear integrity by preventing macromolecules from freely diffusing in or out of the nucleus. Macromolecules smaller than ~40 kDa can passively diffuse through the diffusion barrier, whereas larger macromolecules generally do not. Facilitated transport through NPCs is rapid, adding up to hundreds to thousands of macromolecules per second. Notably, NPCs conduct their cargos in their native state, allowing macromolecules to act immediately after transport, for instance during signal transduction (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019).
Most NPC proteins typically form a symmetric core that possesses an 8-fold rotational symmetry (nucleoporins are incorporated in multiples of eight). This symmetric core surrounds the central transport channel and functions as the scaffold onto which asymmetric nucleoporins attach on the cytoplasmic and nuclear compartments to form structures known as the cytoplasmic filaments and nuclear basket, respectively (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019). One inner ring that is embedded within the nuclear envelope, and two outer rings that reside on the inner or outer nuclear membrane generate the symmetric core itself. The major constituent of the outer rings in the NPC is the coat nucleoporin complex, which serves as a structural scaffold and docking site for other nucleoporins. The nuclear basket, composed of Nup153, Nup50 and Tpr, also serves as a hub for organising nuclear architecture and modulating gene transcription, mRNA processing and export (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019).
The majority of NPC architecture appears to be conserved throughout the Eukaryota and was already established in the last common eukaryotic ancestor (DeGrasse et al., Reference DeGrasse, DuBois, Devos, Siegel, Sali, Field, Rout and Chait2009). However, although the proteins and complexes are rather conserved, their arrangements can differ substantially between cells in the same organism or even within the same cell type at the single cell level (Ori et al., Reference Ori, Banterle, Iskar, Andrés-Pons, Escher, Khanh Bui, Sparks, Solis-Mezarino, Rinner, Bork, Lemke and Beck2013). Specifically, how the NPC connects with the lamina and mRNA transport is likely to be highly divergent between different lineages (Rout et al., Reference Rout, Obado, Schenkman and Field2017).
Proteomics analyses of NPC-containing fractions from T. brucei provided a comprehensive inventory of its nucleoporins, which clearly share a similar fold type, domain organization, composition and modularity in comparison with metazoan and yeast (DeGrasse et al., Reference DeGrasse, DuBois, Devos, Siegel, Sali, Field, Rout and Chait2009). Further, an exhaustive interactome assigned T. brucei nucleoporins to discrete NPC substructures, which despite retaining similar protein composition also presented remarkable architectural differences (Obado et al., Reference Obado, Brillantes, Uryu, Zhang, Ketaren, Chait, Field and Rout2016; illustrated in Fig. 2). Briefly, while most elements of the inner core are conserved, multiple peripheral structures are highly dissimilar, possibly to accommodate divergent nuclear and cytoplasmic functions (Obado et al., Reference Obado, Brillantes, Uryu, Zhang, Ketaren, Chait, Field and Rout2016). TbNPC is highly symmetric, with asymmetry only provided by its two nuclear basket Nups (Obado et al., Reference Obado, Brillantes, Uryu, Zhang, Ketaren, Chait, Field and Rout2016). Further, orthologues of cytoplasmic Nups or mRNA remodelling factors are absent in trypanosomes. Notably, TbNup76, likely the cytoplasm-specific Nup82/88 orthologue, localizes to both faces of the NPC (Obado et al., Reference Obado, Brillantes, Uryu, Zhang, Ketaren, Chait, Field and Rout2016). Overall, trypanosomes present substantial variation in the pore membrane proteins and the absence of critical components involved in mRNA export in fungi and animals. Additionally, there is evidence supporting a Ran-dependent system for mRNA export in trypanosomes, which suggests distinct mechanisms of protein and mRNA transport (Obado et al., Reference Obado, Brillantes, Uryu, Zhang, Ketaren, Chait, Field and Rout2016).
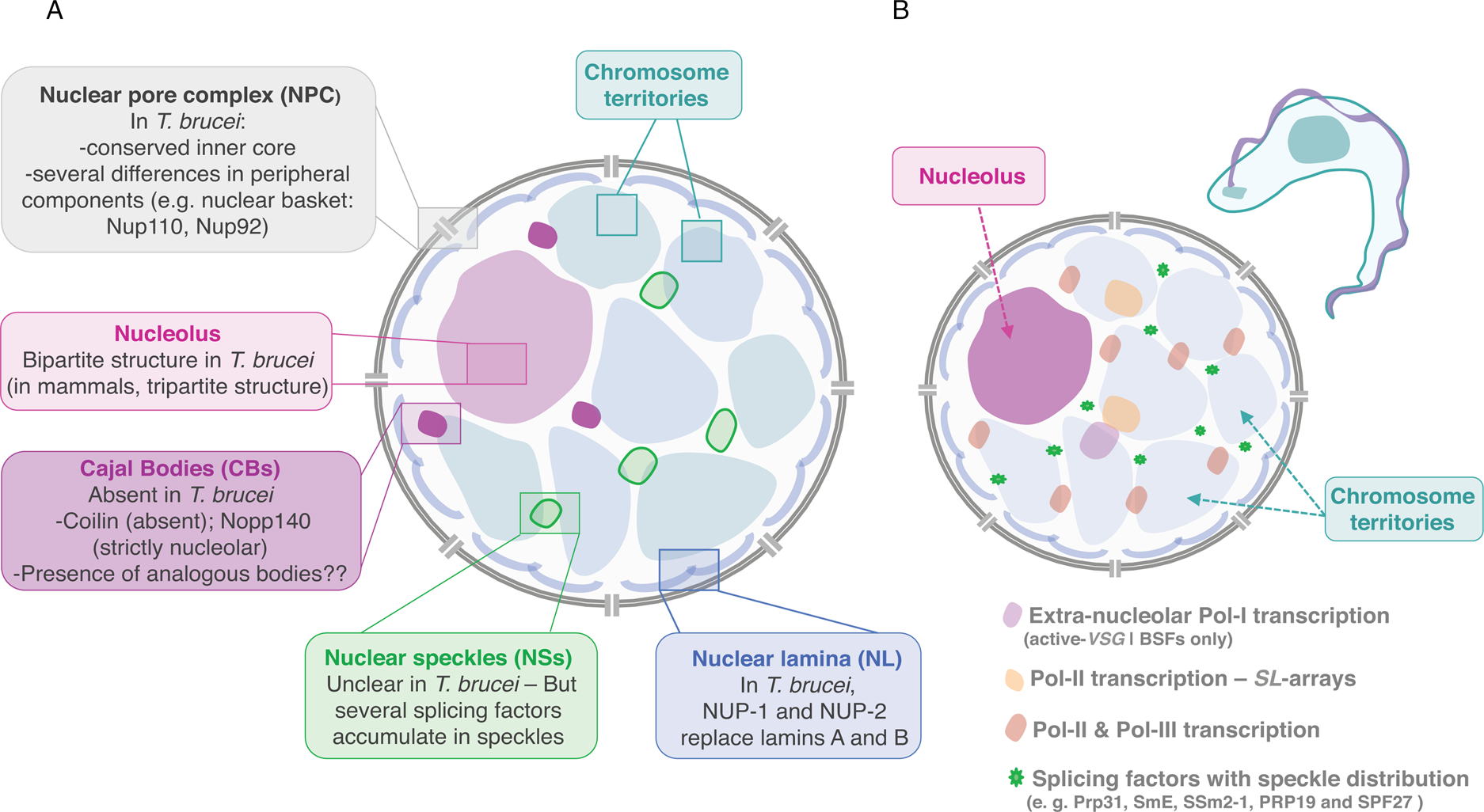
Fig. 2. Nuclear organization and nuclear bodies. (A) The schematics represents the mammalian nucleus, the lateral boxes highlight differences in T. brucei. (B) The schematics represents the nuclear organization and nuclear bodies in T. brucei. Trypanosome-specific compartments are highlighted, such as the bloodstream form (BSF)-specific expression-site body (active-VSG-Expression-Site transcription; extra-nucleolar Pol-I transcription) and the Spliced Leader (SL)-array transcription compartments.
TbNup110 and TbNup92, the two components of the nuclear basket, are predicted to have predominantly coiled-coil structure and are likely to represent the Mlp/Tpr proteins of trypanosomes (Holden et al., Reference Holden, Koreny, Obado, Ratushny, Chen, Chiang, Kelly, Chait, Aitchison, Rout and Field2014). Despite performing similar roles in chromosome segregation, TbNup92 has a restricted taxonomic distribution and appears to have a distinct evolutionary origin than Mlp. Further, unlike Mlp, there was no evidence for a role on the creation of transcriptional boundaries, consistent with trypanosome genome organization and gene expression control (Holden et al., Reference Holden, Koreny, Obado, Ratushny, Chen, Chiang, Kelly, Chait, Aitchison, Rout and Field2014). However, TbNup92-knockout cells differentially expressed genes associated with RNA turnover, raising the interesting possibility that TbNup92 might associate with a particular subset of RNA-binding proteins (Holden et al., Reference Holden, Koreny, Obado, Ratushny, Chen, Chiang, Kelly, Chait, Aitchison, Rout and Field2014).
Notably, in T. brucei as well as related organisms, a comprehensive analysis on whether there are changes in the NPC composition or structure following differentiation into different developmental stages is yet to be performed (Rout et al., Reference Rout, Obado, Schenkman and Field2017); and if such changes occur, whether those play a role in gene expression modulation is yet to be investigated.
Nuclear lamina
In mammals, NL is a meshwork consisting of A- and B-type lamins and lamin-associated proteins, which lines the inner nuclear membrane. In differentiated cells, lamin expression is critical to sustain nuclear architecture, prevent abnormal blebbing of the nuclear envelope, and position the NPCs (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008). NL can influence transcriptional activity and interact with a wide range of transcription factors; it is also involved in the compaction of peripheral chromatin (Shevelyov and Ulianov, Reference Shevelyov and Ulianov2019). Eukaryotic heterochromatin, which is mainly located at the nuclear periphery, is subdivided into densely packed constitutive heterochromatin, including pericentromeric and telomeric chromosomal regions, and the less condensed or so-called facultative heterochromatin located in chromosomal arms (Finn and Misteli, Reference Finn and Misteli2019). Chromosomal regions interacting with the NL are designated lamina-associated domains (LADs) have been identified in a wide-range of eukaryotes, from nematodes to humans, and contain mostly silent or weakly expressed genes (Shevelyov and Ulianov, Reference Shevelyov and Ulianov2019). This supports the idea that NL is a repressive nuclear compartment.
Lamin genes were found in metazoa but appeared to be absent in plants and unicellular organisms. In mammals, two major A-type lamins (lamin A and C) and two major B-type lamins (lamin B1 and B2) have been identified and characterized (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008). They are composed of a long central α-helical rod domain, flanked by globular N-terminal (head) and C-terminal (tail) domains, which self-assemble into higher-order structures whose basic subunit is a coiled-coil dimer (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008). Notably, aberrant lamin protein structure or expression can lead to irregular nuclei and abnormal gene expression. Indeed, hundreds of mutations have been identified in human lamins and linked to diseases, collectively known as laminopathies that include progeria and muscular dystrophies (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008). Interestingly, examples from yeast and plants suggest that alternative, non-lamin, molecular systems can construct an NL (Dechat et al., Reference Dechat, Pfleghaar, Sengupta, Shimi, Shumaker, Solimando and Goldman2008).
In T. brucei, an analogous to vertebrate lamins, NUP-1 is a major component of the nucleoskeleton and plays a key role on heterochromatin organization at the nuclear periphery (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012; illustrated in Fig. 2). NUP-1 is a critical component of a stable network at the inner face of the trypanosome nuclear envelope, its depletion leads to abnormally shaped nuclei and disrupts NPCs and chromosomes organization (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012). NUP-1 affinity purification led to the identification of a second coiled-coil protein, designated NUP-2. Following NUP-2 depletion, NUP-1 is mislocalized and vice versa, strongly suggesting that NUP-1 and NUP-2 form a co-dependent network (Maishman et al., Reference Maishman, Obado, Alsford, Bart, Chen, Ratushny, Navarro, Horn, Aitchison, Chait, Rout and Field2016). NUP-2 knockdown leads to severe fitness cost and a dramatic impact on nuclear architecture including severe changes to the nuclear envelope and chromosomal organization. Moreover, NUP-1 and NUP-2 are conserved across trypanosomes; from a structural and functional perspective, they behave similarly to lamins (Maishman et al., Reference Maishman, Obado, Alsford, Bart, Chen, Ratushny, Navarro, Horn, Aitchison, Chait, Rout and Field2016).
Notably, while the active VSG-ES resides within a transcription factory adjacent to the nucleolus (Navarro and Gull, Reference Navarro and Gull2001) the silent VSG-ESs are located at the extra-nucleolar nucleoplasm but at more peripheral locations in BSFs (Chavez et al., Reference Chaves, Zomerdijk, Dirks-Mulder, Dirks, Raap and Borst1998; Landeira and Navarro, Reference Landeira and Navarro2007; Fig. 2B and Fig. 3). Further, all VSG-ESs localize to the nuclear envelope and appear to form constitutive heterochromatin in insect-stage cells (Landeira and Navarro, Reference Landeira and Navarro2007). This is consistent with the idea that the NL is a repressive compartment. Curiously, in Plasmodium, all silenced var genes localize in a series of clusters at the nuclear periphery, however, the transcription of the active var gene also occurs at a specific site at the nuclear periphery, where the activated gene moves away from the silenced clusters (Duraisingh et al., Reference Duraisingh, Voss, Marty, Duffy, Good, Thompson, Freitas-Junior, Scherf, Crabb and Cowman2005; Freitas-Junior et al., Reference Freitas-Junior, Hernandez-Rivas, Ralph, Montiel-Condado, Ruvalcaba-Salazar, Rojas-Meza, Mâncio-Silva, Leal-Silvestre, Gontijo, Shorte and Scherf2005; Lemieux et al., Reference Lemieux, Kyes, Otto, Feller, Eastman, Pinches, Berriman, Su and Newbold2013).
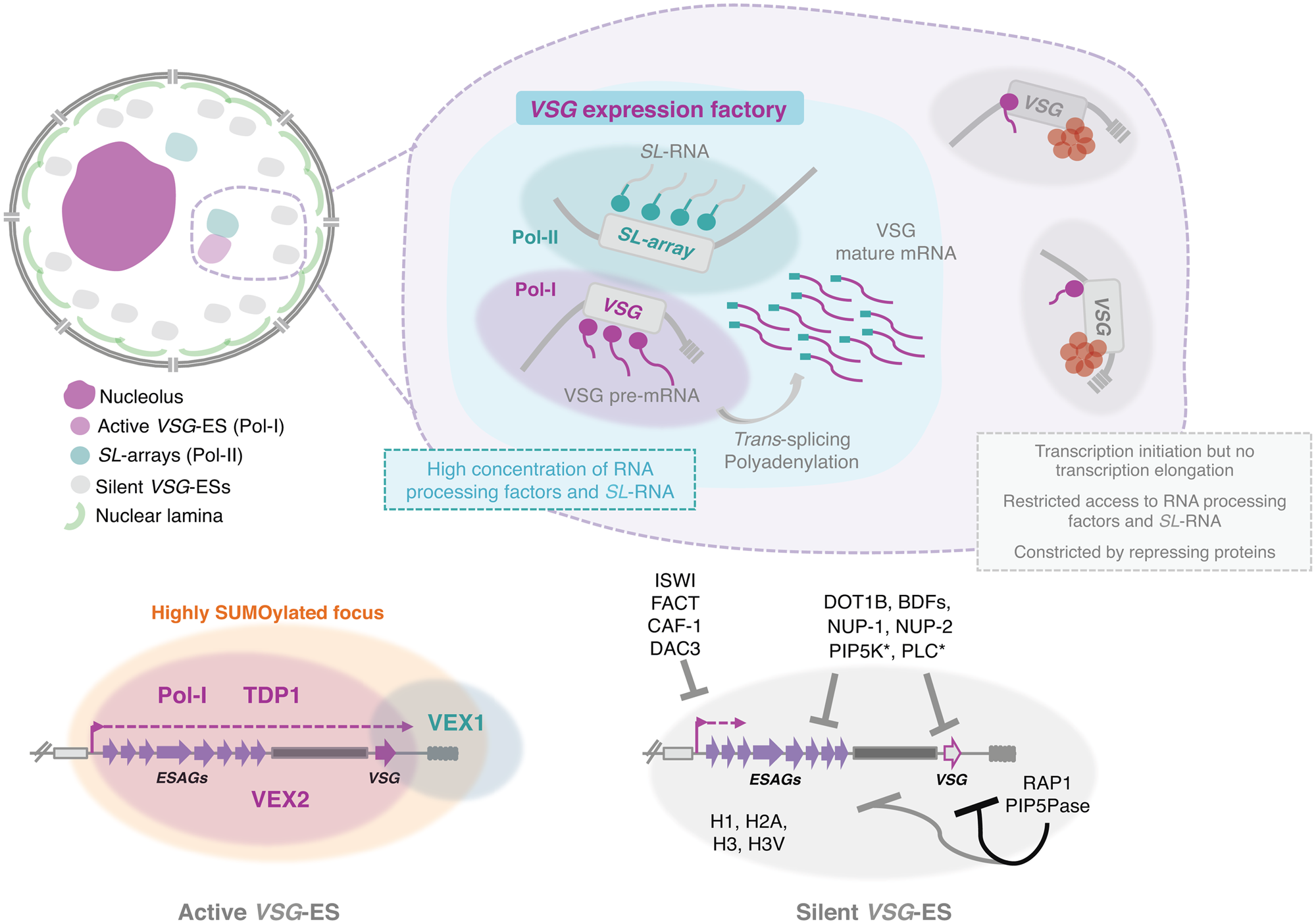
Fig. 3. Nuclear organization and VSG expression in T. brucei bloodstream forms. The single active-VSG establishes a stable inter-chromosomal interaction with one of the SL-arrays. VEX2 orchestrates this spatial integration, which is critical to (1) sustain monogenic expression, (2) enhance RNA processing (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). The active-VSG gene is transcribed at very high levels by Pol-I generating the most abundant protein in the cell. Proximity to the SL-array likely leads to a high local concentration of SL-RNA therefore facilitating trans-splicing. It is possible that several factors associated with RNA processing (splicing, polyadenylation, etc.) are concentrated in this sub-nuclear compartment as well. The SL-array appears to function as a post-transcriptional enhancer and such control might extend beyond VSG genes (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). The active VSG-ES lies within a highly SUMOylated focus (López-Farfán et al., Reference López-Farfán, Bart, Rojas-Barros and Navarro2014); TDP1 is a high mobility group box protein that facilitates Pol-I transcription and is enriched at the active-ES (Narayanan and Rudenko, Reference Narayanan and Rudenko2013). VEX2 and VEX1 form discrete protein condensates that associate with the active-VSG and the SL-array, respectively. The VEX complex, especially VEX2, sustains the exclusive interaction between a single VSG-ES and the SL-array; following its depletion, all VSG-ESs can access the SL-arrays and are derepressed (Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019, Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). The silent VSG-ESs have more peripheral locations; transcription by Pol-I is initiated at the same rate as at the active-locus but transcription elongation is unsuccessful; these sites also have restricted access to RNA processing factors and substrates (Vanhamme et al., Reference Vanhamme, Poelvoorde, Pays, Tebabi, Van Xong and Pays2000; Kassem et al., Reference Kassem, Pays and Vanhamme2014); several repressing factors associated with heterochromatin formation (red circles) sustain their inactive state. For instance, ISWI (Hughes et al., Reference Hughes, Wand, Foulston, Young, Harley, Terry, Ersfeld and Rudenko2007), FACT (Denninger and Rudenko, Reference Denninger and Rudenko2014), CAF-1 (Alsford and Horn, Reference Alsford and Horn2012) or DAC3 (Wang et al., Reference Wang, Kawahara and Horn2010) repress transcription near the promoter in silent ESs. Telomeric ES proteins, such as RAP1 (Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009) or PIP5Pase (Cestari et al., Reference Cestari, McLeland-Wieser and Stuart2019), repress transcription of the whole ES, the repressive gradient is stronger near the telomeres (indicated by the darker line). Other repressive proteins include DOT1B (Figueiredo et al., Reference Figueiredo, Janzen and Cross2008), bromodomain proteins (BDFs) (Schulz et al., Reference Schulz, Mugnier, Paulsen, Kim, Chung, Tough, Rioja, Prinjha, Papavasiliou and Debler2015) and PIP5K and PLC, marked with an asterisk because they are the only ones that do not localize to the nucleus (Cestari and Stuart, Reference Cestari and Stuart2015). Moreover, the integrity of the nuclear lamina is critical to maintain this repressive state (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012).
In T. brucei, NUP-1 plays a role on epigenetic control of developmentally regulated loci. Indeed, following NUP-1 knockdown, megabase chromosome telomeres reposition, multiple VSG-ESs become active, and the frequency of VSG switching increases (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012; Rout et al., Reference Rout, Obado, Schenkman and Field2017). Additionally, the active VSG-ES promoter fails to migrate to the nuclear periphery upon differentiation, and metacyclic VSGs are derepressed in insect-stage cells, both likely associated with the defective formation and/or maintenance of a repressive heterochromatin compartment (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012). Heterochromatin-based silencing in trypanosomes involves several proteins, such as ISWI, RAP1 and histone deacetylase (DAC) 3 (Hughes et al., Reference Hughes, Wand, Foulston, Young, Harley, Terry, Ersfeld and Rudenko2007; Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009; Wang et al., Reference Wang, Kawahara and Horn2010), whilst histone H1 participates in maintaining condensed chromatin in silenced regions (Povelones et al., Reference Povelones, Gluenz, Dembek, Gull and Rudenko2012). Strikingly, T. brucei lacks H3K9me3, a well-characterized marker for heterochromatin, and heterochromatin-protein 1 (HP1) (Berriman et al., Reference Berriman2005). It is noteworthy that the misregulation of VSG and procyclin genes is quite modest following NUP-1 depletion (up to 10-fold) (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012); however, it demonstrates that NUP-1 and the trypanosome NL integrate a series of possibly multiple mechanisms that constrain the inactive VSG-ESs and reinforce their silent state.
Membraneless nuclear bodies
Eukaryotic cells contain membraneless organelles, designated cellular bodies, which compartmentalize essential biochemical reactions and cellular functions. These bodies are generated by phase separation mediated by cooperative interactions between multivalent molecules (Strom and Brangwynne, Reference Strom and Brangwynne2019; Razin and Gavrilov, Reference Razin and Gavrilov2020). Well-characterized examples of such organelles in the nucleus are nucleoli, which are sites of rRNA biogenesis; Cajal bodies (CB), which are assembly sites for small nuclear ribonucleoproteins (RNPs); and nuclear speckles (NSs), which are storage compartments for RNA processing factors (Strom and Brangwynne, Reference Strom and Brangwynne2019; Razin and Gavrilov, Reference Razin and Gavrilov2020). Besides their ability to move throughout the nucleus, another fascinating feature of several nuclear bodies is their ability to form within the nuclear milieu without apparent support structures, again consistently with a self-organization model. Moreover, these organelles exhibit properties similar to liquid droplets, being able to undergo fission and fusion. In fact, mixtures of specific RNA and certain RNA-binding proteins are able to form phase-separated bodies in vitro (Guo et al., Reference Guo, Manteiga, Henninger, Sabari, Dall'Agnese, Hannett, Spille, Afeyan, Zamudio, Shrinivas, Abraham, Boija, Decker, Rimel, Fant, Lee, Cisse, Sharp, Taatjes and Young2019; Hondele et al., Reference Hondele, Sachdev, Heinrich, Wang, Vallotton, Fontoura and Weis2019).
Nucleolus
The nucleolus is likely to be the most distinctive nuclear compartment, certainly the largest and the site of ribosome biogenesis where the 45S ribosomal repeats are clustered. Indeed, pre-rRNA transcription and processing as well as the assembly of the 40S and 60S complexes take place in this nuclear body (Hernandez-Verdun et al., Reference Hernandez-Verdun, Roussel, Thiry, Sirri and Lafontaine2010). In animals and plants, the nucleolus presents a tripartite substructure, which can be observed by electron microscopy. This tripartite substructure includes fibrillar centres (FC) surrounded by a dense fibrillar component (DFC); both embedded in the granular component, the biggest nucleolar subdomain composed of RNP granules. FC stores inactive rRNA genes, whereas DFC is electron dense given the high concentration of RNPs and is involved in early rRNA processing (Hernandez-Verdun et al., Reference Hernandez-Verdun, Roussel, Thiry, Sirri and Lafontaine2010).
Eukaryotic ribosomes are composed of 18S, 5.8S, 28S and 5S rRNA subunits and approximately 80 associated proteins. The four rRNA molecules are the main structural and catalytic components of the ribosome. In most eukaryotes, genes encoding for 18S, 5.8S, 28S are organized in tandem repeats, which are transcribed by RNA Polymerase I (Pol-I) into a primary transcript further processed into the mature 18S, 5.8S, 28S rRNAs (Hernandez-Verdun et al., Reference Hernandez-Verdun, Roussel, Thiry, Sirri and Lafontaine2010). Transcription occurs in the boundary between FC and DFC. 5S rRNA genes, on the other hand, are transcribed in the nucleoplasm by Pol-III (Hernandez-Verdun et al., Reference Hernandez-Verdun, Roussel, Thiry, Sirri and Lafontaine2010).
Similarly to other eukaryotes, the nucleolus is the most distinctive membraneless sub-nuclear body in trypanosomes and Leishmania parasites that can be easily observed by light and electron microscopy (Ogbadoyi et al., Reference Ogbadoyi, Ersfeld, Robinson, Sherwin and Gull2000; Nepomuceno-Mejía et al., Reference Nepomuceno-Mejía, Lara-Martínez, Cevallos, López-Villaseñor, Jiménez-García and Hernández2010). Presently, FCs have not been identified in the nucleolus of these organisms, which presents a bipartite structure, similarly to other protozoa, yeast, invertebrates, fish and amphibians (Ogbadoyi et al., Reference Ogbadoyi, Ersfeld, Robinson, Sherwin and Gull2000; Nepomuceno-Mejía et al., Reference Nepomuceno-Mejía, Lara-Martínez, Cevallos, López-Villaseñor, Jiménez-García and Hernández2010; illustrated in Fig. 2). Unlike more complex eukaryotes, during cell division in T. brucei, the nuclear envelope is preserved, chromatin does not condense and the nucleolus does not disassemble. As mitosis progresses, the nucleolus stretches, is pulled via the spindle fibres to opposite poles of the nucleus and ultimately divided into two independent structures (Ogbadoyi et al., Reference Ogbadoyi, Ersfeld, Robinson, Sherwin and Gull2000). This process occurs in the absence of intermediate structures such as prenucleolar bodies, found in other organisms (Hernandez-Verdun et al., Reference Hernandez-Verdun, Roussel, Thiry, Sirri and Lafontaine2010).
In T. brucei and similarly to other organisms, the biogenesis of ribosome subunits starts in the nucleolus and ends in the cytoplasm. The 5S rRNA is imported to the nucleolus very early in the biogenesis process and incorporated into the 90S pre-ribosome as an RNP complex; it later undergoes spatial rearrangement to facilitate subsequent maturation steps of the 60S subunit (Prohaska and Williams, Reference Prohaska and Williams2009; Liu et al., Reference Liu, Gutierrez-Vargas, Wei, Grassucci, Ramesh, Espina, Sun, Tutuncuoglu, Madison-Antenucci, Woolford, Tong and Frank2016). The pre-60S particle is translocated from the nucleus to the cytoplasm through interactions between P34 and P37and exportin 1 and Nmd3, as well as r-proteins uL3 and uL11 (Prohaska and Williams, Reference Prohaska and Williams2009). The biogenesis of the 40S subunit in T. brucei occurs very similar to what has been described in yeast (Ferreira-Cerca et al., Reference Ferreira-Cerca, Pöll, Kühn, Neueder, Jakob, Tschochner and Milkereit2007). Interestingly, this subunit contains a trypanosomatid-specific helical structure that has been proposed to participate in translation initiation by interacting with the SL-sequence and its unusually modified cap (Hashem et al., Reference Hashem, des Georges, Fu, Buss, Jossinet, Jobe, Zhang, Liao, Grassucci, Bajaj, Westhof, Madison-Antenucci and Frank2013).
In humans, the nucleolus has been associated with multiple functions that extend beyond ribosome biogenesis, one being a cellular stress sensor (Rubbi and Milner, Reference Rubbi and Milner2003). Studies in trypanosomes suggest this may be the case in trypanosomatid parasites as well (Elias et al., Reference Elias, Marques-Porto, Freymüller and Schenkman2001; Barquilla et al., Reference Barquilla, Crespo and Navarro2008). Moreover, the nucleolus appears as a largely self-organized structure. Indeed, its integrity relies on both active Pol-I transcription and high interactivity between ribosomal components (Raska et al., Reference Raska, Shaw and Cmarko2006). Interestingly, ectopic expression of rRNA leads to the formation of micronucleoli in Drosophila (Karpen et al., Reference Karpen, Schaefer and Laird1988), again consistently with a model of self-organized nuclear compartmentalization. In trypanosomes, specifically, depletion of Pol-I-specific subunits leads to abnormal nucleoli (Devaux et al., Reference Devaux, Kelly, Lecordier, Wickstead, Perez-Morga, Pays, Vanhamme and Gull2007) and depletion of TOR1 kinase leads to Pol-I and nucleolar dispersion, most likely as a consequence of Pol-I transcription inhibition (Barquilla et al., Reference Barquilla, Crespo and Navarro2008). In T. cruzi, development from a proliferative to non-proliferative stage, which is associated with a pronounced drop in transcriptional activity, is also accompanied by nucleolar dispersion (Elias et al., Reference Elias, Marques-Porto, Freymüller and Schenkman2001).
Further details on nucleolar structure and function in trypanosomatid parasites have been recently reviewed by (Martínez-Calvillo et al., Reference Martínez-Calvillo, Florencio-Martínez and Nepomuceno-Mejía2019).
Nuclear speckles and Cajal bodies
In complex eukaryotes such as animals and plants, CBs are involved in the post-transcriptional maturation of small nuclear (snRNAs) and small nucleolar RNAs (snoRNAs) and the biogenesis of nuclear RNPs, including some nucleolar proteins, snoRNPs and snRNPs (Sawyer et al., Reference Sawyer, Sturgill, Sung, Hager and Dundr2016). The number of CBs varies across cell types and at a single-cell level within the same cell type (in mammalian cells typically 0–10 CBs per nucleus, ranging 0.1–2 μm in diameter). CBs are more abundant in cells with high transcriptional activity and are highly dynamic but structurally stable structures. They continuously exchange components into and out of the domain in response to changes in the cellular environmental (Sawyer et al., Reference Sawyer, Sturgill, Sung, Hager and Dundr2016). Interestingly, components of the SNAPc complex were reported to be enriched within the CB, suggesting a strong link between snRNA gene transcription and CBs. Several studies also indicate that CBs influence the levels and processivity of factors crucial for efficient RNA splicing; indeed CBs may influence splicing kinetics through different pathways (Sawyer et al., Reference Sawyer, Sturgill, Sung, Hager and Dundr2016).
Coilin and the nucleolar protein Nopp140 are the two key markers of CBs. Coilin has been implicated in the link between the nucleolus and CBs; indeed CBs are frequently detected at the nucleolar periphery and even within nucleoli. Coilin is a key structural component of CBs, is involved in RNP metabolism within these nuclear bodies and it also appears to have a role on general chromatin organization (Machyna et al., Reference Machyna, Neugebauer and Staněk2015). Its N-terminal domain is responsible for the self-oligomerization activity, truncation or mutation of phosphorylation sites in the conserved C-terminal region leads to a dramatic alteration in the number of CBs (Shpargel et al., Reference Shpargel, Ospina, Tucker, Matera and Hebert2003). On the other hand, Nopp140 does not localize strictly to CBs and it appears to serve generally as a chaperone for RNPs; it moves between the nucleolus and the CBs, but also between the nucleolus and the cytoplasm (Isaac et al., Reference Isaac, Yang and Meier1998). Indeed, it not only interacts with coilin, but also associates with several nucleolar proteins (Isaac et al., Reference Isaac, Yang and Meier1998).
Trypanosoma brucei appears to lack a coilin homologue and TbNopp140 is strictly nucleolar, strongly suggesting that CBs, in the strict sense of the definition, are absent in these parasites (Berriman et al., Reference Berriman2005; Kelly et al., Reference Kelly, Singleton, Wickstead, Ersfeld and Gull2006) (Fig. 2). Additionally, T. brucei possesses two homologues of Nopp140, a canonical Nopp140 and a Nopp140-like protein, both are phosphorylated and co-immunoprecipitate with Pol-I and might play a role on nucleoplasmic snoRNPs shuttling (Kelly et al., Reference Kelly, Singleton, Wickstead, Ersfeld and Gull2006). Given the absence of CBs, it has been proposed that in T. brucei RNPs are probably assembled in analogous bodies: a possible candidate was a compartment identified as Spliced-leader-associated RNA (SLA1)-containing subnuclear site that did not colocalize with SL-RNA (Hury et al., Reference Hury, Goldshmidt, Tkacz and Michaeli2009). SLA1 guides the pseudouridylation at position −12 (relative to the 5′ splice site) of the SL-RNA in all trypanosomatid species.
NSs or splicing speckles were originally discovered as sites for splicing factor storage and modification and were later revealed to play a general role in RNA metabolism. Subsequently, numerous proteins involved in epigenetic regulation, chromatin organization, DNA repair and RNA modifications were found in NSs (Galganski et al., Reference Galganski, Urbanek and Krzyzosiak2017). Similar to other membraneless bodies with liquid-like properties, NSs are characterized by the dynamic exchange of components within the nucleoplasm, sharing some proteins with other nuclear bodies (Galganski et al., Reference Galganski, Urbanek and Krzyzosiak2017).
In trypanosomes, trans-splicing occurs for every single mRNA, there are only two known cis-spliced introns in T. brucei; both mechanisms seem to require the spliceosome. Notably, trypanosomes encode for all snRNA and many spliceosomal proteins described in other eukaryotes but also encode for a few specific factors (Palfi et al., Reference Palfi, Lücke, Lahm, Lane, Kruft, Bragado-Nilsson, Séraphin and Bindereif2000; Ambrósio et al., Reference Ambrósio, Lee, Panigrahi, Nguyen, Cicarelli and Günzl2009; Preusser et al., Reference Preusser, Palfi and Bindereif2009; also reviewed by Günzl, Reference Günzl2010; Michaeli, Reference Michaeli2011 and Clayton, Reference Clayton2019). Interestingly, splicing factors such as Prp31, SmE, SSm2-1, PRP19 and SPF27 have a speckle-like organization and appear to be compartmentalized in specific nuclear areas (Liang et al., Reference Liang, Liu, Liu, Tschudi and Michaeli2006; Tkacz et al., Reference Tkacz, Lustig, Stern, Biton, Salmon-Divon, Das, Bellofatto and Michaeli2007; Reference Tkacz, Gupta, Volkov, Romano, Haham, Tulinski, Lebenthal and Michaeli2010; Ambrósio et al., Reference Ambrósio, Badjatia and Günzl2015; illustrated in Fig. 2B).
Recent advances in more complex eukaryotes suggested that NSs facilitate integrated regulation of gene expression (Galganski et al., Reference Galganski, Urbanek and Krzyzosiak2017). A substantial fraction of the mammalian genome is preferentially organized around nuclear bodies such as the nucleolus and NSs; these bodies have been proposed to act as inter-chromosomal hubs that shape the overall packaging of DNA in the nucleus (Quinodoz et al., Reference Quinodoz, Ollikainen, Tabak, Palla, Schmidt, Detmar, Lai, Shishkin, Bhat, Takei, Trinh, Aznauryan, Russell, Cheng, Jovanovic, Chow, Cai, McDonel, Garber and Guttman2018). Additionally, many active genes reproducibly position near NSs, but the nature of such associations had remained unclear until recently, when a study linked them to stochastic gene expression amplification (Kim et al., Reference Kim, Venkata, Hernandez Gonzalez, Khanna and Belmont2020). Whether similar associations are present and play a role in genome organization and gene expression in trypanosomes and related organisms remains to be explored.
In summary, compartmentalization within the nucleoplasm enables functional specialization; in fact, key nuclear functions such as transcription or RNA processing are not homogeneously distributed throughout the nucleus. In the next chapter, I will specifically cover the current knowledge on transcription regulation and compartmentalization in trypanosomes.
Transcription regulation
To our knowledge, all trypanosomatids employ primarily polycistronic transcription, where multiple open reading frames with no functional association are transcribed in tandem. Evidence suggests that the position within the PTU is associated with messenger RNA (mRNA) copy number (Kelly et al., Reference Kelly, Kramer, Schwede, Maini, Gull and Carrington2012). The nascent RNAs are processed into mature mRNAs, through a combination of trans-splicing and polyadenylation (reviewed by Günzl, Reference Günzl2010; Michaeli, Reference Michaeli2011; Clayton, Reference Clayton2019). Notably, mature mRNAs bear an unusual hypermethylated 5′ cap structure (Bangs et al., Reference Bangs, Crain, Hashizume, McCloskey and Boothroyd1992). The genome is therefore constitutively transcribed and mRNA abundance is primarily controlled at the post-transcriptional level in striking contrast with more complex eukaryotes, where a specific promoter usually regulates the transcription of each gene (Koumandou et al., Reference Koumandou, Natesan, Sergeenko and Field2008).
Exceptions to this mechanism are the genomic loci encoding for highly abundant surface-exposed antigens, VSGs and procyclins: these loci are transcribed at very high levels by Pol-I and not Pol-II, which transcribes the majority of PTUs. Both VSGs and procyclins expression is developmentally regulated, the former expressed in the mammalian-stage and the latter in the insect-stage (Navarro et al., Reference Navarro, Peñate and Landeira2007; Daniels et al., Reference Daniels, Gull and Wickstead2010).
RNA polymerase I (Pol-I)
Trypanosoma brucei is the only organism known to have evolved a multifunctional Pol-I system that is used for rRNA synthesis and for the expression of highly abundant antigens (Günzl et al., Reference Günzl, Bruderer, Laufer, Schimanski, Tu, Chung, Lee and Lee2003). As previously mentioned, VSGs and procyclins are strongly developmentally regulated and therefore Pol-I transcription in T. brucei is intimately linked to differentiation between different life cycle stages as well as antigenic variation in the mammalian host, which is critical to sustain persistent infections.
In the vast majority of eukaryotes, Pol-I is recruited to simple promoters, which contain an upstream element located 100 bp from the transcription start site. Such promoters are exclusively used for rRNA gene expression, specifically the 45S rRNA precursor, further processed into 18S, 5.8S and 28S rRNA. Two protein complexes, the selectivity factor 1 (SL1) and the upstream binding factor (UBF), are essential for Pol-I recruitment to the rRNA promoter (Russell and Zomerdijk, ). The interaction between Pol-I and SL1 is mediated by a single polypeptide named RRN3 in humans; a UBF dimer is further required to activate rRNA transcription. In yeast, RRN3 is conserved, whereas the three subunits of the core factor (the functional equivalent of SL1) and the six UBF subunits share no sequence similarity with the mammalian counterparts (Russell and Zomerdijk, ). Recent CryoEM studies suggest that, unlike the Pol-II system, promoter specificity relies on a distinct ‘bendability’ and ‘meltability’ of the promoter sequence that enables contacts between initiation factors, DNA and polymerase (Engel et al., Reference Engel, Gubbey, Neyer, Sainsbury, Oberthuer, Baejen, Bernecky and Cramer2017). In eukaryotic cells, although the number of rRNA genes is much lower than the number of protein-coding genes, Pol-I transcription usually accounts for more than 50% of the total transcriptional activity, which results from impressively high transcription initiation rates. Notably, mammalian Pol-I is unable to synthesize functional mRNA (Russell and Zomerdijk, ).
Similarly to all eukaryotes, T. brucei Pol-I transcribes the 45S rRNA precursor in the nucleolus; however, it also transcribes procyclins and VSGs mRNAs from perinucleolar and extra-nucleolar locations, respectively (Navarro and Gull, Reference Navarro and Gull2001) (Figs 2B and 3). The rRNA, VSG and procyclin gene promoters are structurally different, suggesting that they recruit different transcription factors. Since the last two promoters are absent in related organisms T. cruzi and Leishmania spp., one would expect to find T. brucei-specific proteins for VSG and procyclin gene transcription.
In T. brucei, both bioinformatics and biochemical analyses have unravelled 10 out of 12 Pol-I subunits: RPA1, RPA2, RPC40, RPB5z, RPB6z, RPB8, RPC19, RPB10z and RPA12 (Walgraffe et al., Reference Walgraffe, Devaux, Lecordier, Dierick, Dieu, Van den Abbeele, Pays and Vanhamme2005; Nguyen et al., Reference Nguyen, Schimanski, Zahn, Klumpp and Günzl2006). RPB5z, RPB6z and RPB10z are RPB5, RPB6 and RPB10 paralogs, respectively. Further, T. brucei presents functional diversification of isoforms that are conventionally shared RNA polymerase subunits (Devaux et al., Reference Devaux, Kelly, Lecordier, Wickstead, Perez-Morga, Pays, Vanhamme and Gull2007). Trypanosoma brucei also has a specific component (or a divergent orthologue of yeast RPA43), RPA31, which is critical for Pol-I transcription and cell viability (Walgraffe et al., Reference Walgraffe, Devaux, Lecordier, Dierick, Dieu, Van den Abbeele, Pays and Vanhamme2005; Nguyen et al., Reference Nguyen, Schimanski and Günzl2007). Although conceivable, there is no evidence that RPA31, RPB5z, RPB6z and RPB10z play a specific role on mRNA transcription by Pol-I in T. brucei.
The class I transcription factor A (CITFA) has been identified in T. brucei; its purification led to the identification of seven novel subunits, termed CITFA-1 to -7, plus the dynein light chain DYNLL1 (also known as LC8) (Brandenburg et al., Reference Brandenburg, Schimanski, Nogoceke, Nguyen, Padovan, Chait, Cross and Günzl2007; Nguyen et al., Reference Nguyen, Nguyen, Lee, Panigrahi and Günzl2012). CITFA binds rRNA, VSG and procyclin promoters and therefore is a general Pol-I transcription factor in T. brucei; its depletion is unsurprisingly lethal (Brandenburg et al., Reference Brandenburg, Schimanski, Nogoceke, Nguyen, Padovan, Chait, Cross and Günzl2007). Further, TDP1, a high motility group box containing protein, which facilitates Pol-I transcription, is highly enriched at the active VSG-ES (compared to silent) and in the nucleolus; a blockade in TDP1 synthesis results in a pronounced reduction of Pol-I-derived transcripts (Narayanan and Rudenko, Reference Narayanan and Rudenko2013). TDP1 overexpression was sufficient to open the chromatin of silent VSG-ESs and disrupt VSG monogenic expression (Aresta-Branco et al., Reference Aresta-Branco, Sanches-Vaz, Bento, Rodrigues and Figueiredo2019). Moreover, ELP3B was identified as a specific negative regulator of rRNA transcription (no impact on VSG transcription); these observations extend the roles of the Elp3-related proteins to Pol-I transcription units, as they are usually associated with Pol-II transcription in humans and yeast (Alsford and Horn, Reference Alsford and Horn2011).
Notably, all proteins involved in T. brucei Pol-I transcription identified so far are conserved among all trypanosomatids, suggesting that they fulfil general Pol-I functions (Walgraffe et al., Reference Walgraffe, Devaux, Lecordier, Dierick, Dieu, Van den Abbeele, Pays and Vanhamme2005; Nguyen et al., Reference Nguyen, Schimanski, Zahn, Klumpp and Günzl2006; Devaux et al., Reference Devaux, Kelly, Lecordier, Wickstead, Perez-Morga, Pays, Vanhamme and Gull2007). However, it is entirely possible that these common factors evolved specific functions for protein-coding gene transcription in T. brucei. Nevertheless, how T. brucei Pol-I acquired the ability to transcribe mRNA remains mysterious.
RNA polymerase II (Pol-II)
Pol-II synthesizes pre-mRNAs and U-rich short nuclear RNAs (snRNAs). The latter form the core of the spliceosome, involved in processing pre-mRNAs into mature mRNAs. In both cases, the 5′ ends are capped, which requires adding m7G to the 5′ triphosphate end of the primary transcript and takes place co-transcriptionally (Proudfoot et al., Reference Proudfoot, Furger and Dye2002). There are several other co-transcriptional activities, which are assigned to specific subunits or domains within these subunits (Proudfoot et al., Reference Proudfoot, Furger and Dye2002). Trypanosoma brucei Pol-II produces both pre-mRNAs and the spliced leader (SL)-RNA, the latter is detrimental for trans-splicing.
Eukaryotic Pol-II enzymes usually contain 12 subunits, designated RPB1 to RPB12. Specifically, RPB1, RPB2, RPB3 and RPB11 are considered the functional and structural core subunits. Additionally, RPB4 to RPB10 and RPB12 usually contribute to Pol-II ability to respond to activators and tightly bind promoter regions (Proudfoot et al., Reference Proudfoot, Furger and Dye2002). The 12 Pol-II subunits could be identified in T. brucei; RPB1, RPB2, RPB3 and RPB11 were also considered the functional and structural core (Das et al., Reference Das, Li, Liu and Bellofatto2006; Devaux et al., Reference Devaux, Lecordier, Uzureau, Walgraffe, Dierick, Poelvoorde, Pays and Vanhamme2006). Interestingly, trypanosomes have two isoforms of RPB5 and RPB6 (Das et al., Reference Das, Li, Liu and Bellofatto2006; Devaux et al., Reference Devaux, Lecordier, Uzureau, Walgraffe, Dierick, Poelvoorde, Pays and Vanhamme2006). RPB1 is the largest subunit in the T. brucei enzyme and also the most fascinating (Evers et al., Reference Evers, Hammer, Köck, Jess, Borst, Mémet and Cornelissen1989). One of the most remarkable characteristics is the non-structured carboxyl end of the polypeptide, which deviates from the heptapeptide repeat of YSPTSPS of varying length that is characteristic of yeast and mammalian proteins. This repeat is generally involved in the modulation of multiple co-transcriptional processes that include capping, splicing, elongation, polyadenylation and nuclear export, through coordinated kinetic alterations in the phosphorylation of its serines and threonines (Proudfoot et al., Reference Proudfoot, Furger and Dye2002). Despite being non-repetitive, the trypanosome carboxyterminal is phosphorylated and essential for transcription (Evers et al., Reference Evers, Hammer, Köck, Jess, Borst, Mémet and Cornelissen1989). Trypanosoma brucei cdc2-related kinase 9 (CRK9) was found to be responsible for RPB1 phosphorylation, however, surprisingly, when silencing CRK9, there was no impact on Pol-II transcription or co-transcriptional m7G capping. Instead it led to a block of trans-splicing caused by hypomethylation of the SL-RNA unique cap4 (Badjatia et al., Reference Badjatia, Ambrósio, Lee and Günzl2013).
In many organisms, a crucial regulatory point of gene expression is transcription initiation, which requires the formation of a pre-initiation complex that includes multiple proteins that interact with Pol-II. Such transcription factors include TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH, which recruit and position Pol-II at promoter sequences (Hahn, Reference Hahn2004). The only canonical Pol-II promoter in T. brucei is the SL-RNA promoter. In this organism, the identification of general transcription factors was challenged by their extremely divergent amino acid sequences from those of their eukaryotic counterparts. The first transcription factor purified and characterized was a trimeric SNAPc that formed a larger complex with TATA-binding protein, the small subunit of TFIIA (TFIIA2), and a sixth protein (TFIIA1) (Das et al., Reference Das, Zhang, Palenchar, Chatterjee, Cross and Bellofatto2005; Schimanski et al., Reference Schimanski, Nguyen and Günzl2005). This was followed by the identification of TFIIB, TFIIH, TFIIE; later, a TFIIH-associated complex of nine subunits was discovered, and despite exhibiting no motif or sequence conservation that could reveal its identity, it structurally resembled the head module of the much larger mediator complex of other eukaryotes (Schimanski et al., Reference Schimanski, Brandenburg, Nguyen, Caimano and Günzl2006; Lee et al., Reference Lee, Nguyen, Schimanski and Günzl2007, Reference Lee, Cai, Panigrahi, Dunham-Ems, Nguyen, Radolf, Asturias and Günzl2010). More recently, a TFIIF-like or TFL complex has been identified, strongly indicating that trypanosomatids possess a full set of RNA Pol-II general transcription factors, only very divergent from their mammalian and yeast counterparts (Srivastava et al., Reference Srivastava, Badjatia, Lee, Hao and Günzl2018). All these factors are required for SL-RNA transcription and trypanosome viability, but their role, if any, on the transcription of protein-coding genes remains unknown.
In T. brucei, ubiquitously expressed genes lack well-defined Pol-II promoter motifs, with the exception of the spliced-leader RNA promoter. Indeed, the so-called Pol-II disperse promoters lack conserved sequence motifs and tight regulation; however, they are defined by specific chromatin structures. In T. brucei for instance, GT-rich promoters were recently proposed to drive transcription and promote the targeted deposition of the histone variant H2A.Z, showing that even highly dispersed, unregulated promoters might contain specific DNA elements that are able to induce transcription (Wedel et al., Reference Wedel, Förstner, Derr and Siegel2017).
Additionally, Pol-II transcription termination is a tightly regulated process and critical to prevent the elongating Pol-II complex from interfering with the transcription of downstream genes. In kinetoplastid flagellates, the modified base β-D-glycosyl-hydroxymethyluracil (J) replaces a small percentage of thymine residues, mostly in telomeric regions and is synthesized at the DNA level via the precursor 5-hydroxymethyluracil. In T. brucei for instance, base J is exclusively present in the BSF. Notably, in T. brucei and Leishmania major, base J and H3.V are enriched at sites involved in Pol-II termination. Loss of base J and H3.V led to transcription read-through (Reynolds et al., Reference Reynolds, Hofmeister, Cliffe, Siegel, Anderson, Beverley, Schmitz and Sabatini2016; Schulz et al., Reference Schulz, Zaringhalam, Papavasiliou and Kim2016). Recently, a novel base J-binding protein complex involved in Pol-II transcription termination has been identified (Kieft et al., Reference Kieft, Zhang, Marand, Moran, Bridger, Wells, Schmitz and Sabatini2020).
Overall, trypanosomes appear to have limited control over Pol-II transcription initiation, and therefore most of the gene expression control is thought to be post-transcriptional.
RNA polymerase III (Pol-III)
Pol-III is responsible for the transcription of a number of small non-coding RNAs that play a role in translation (tRNA and 5S rRNAs) and other cellular processes (7SL RNA). In T. brucei, tRNA genes can be found widely spread throughout large directional gene clusters on megabase chromosomes, 5S rRNA genes are clustered in chromosome 8 (Berriman et al., Reference Berriman2005).
Expression factories
The SL-RNA expression factory
Given that SL-RNA must be added to the 5′ end of every single mRNA in T. brucei, trans-splicing relies on large quantities of SL transcripts generated by Pol-II transcription from a diploid tandem-repeat locus. Indeed, Pol-II largest subunit is highly concentrated at the SL-RNA genomic loci (illustrated in Fig. 2B). In T. cruzi and Leishmania tarentolae, a single focus is observed possibly due to pairing of both alleles (Dossin and Schenkman, Reference Dossin Fde and Schenkman2005). In contrast, in T. brucei, two distinct foci could be detected in G1 cells indicating that the two SL-arrays occupy distinct chromosome territories (Uzureau et al., Reference Uzureau, Daniels, Walgraffe, Wickstead, Pays, Gull and Vanhamme2008). In T. cruzi, the Pol-II focus disperses following treatment with transcription inhibitors (Dossin and Schenkman, Reference Dossin Fde and Schenkman2005), suggesting that the high concentration and organization of Pol-II around the SL-arrays depends on active-transcription and therefore is not a predefined nuclear structure.
Moreover, SL-RNA transcripts concentrate in a nuclear area that colocalizes with the snRNP protein SmE and SLA1 RNA, an RNA involved in the SL-RNA modification. This strongly suggests that there is a spatially defined SL-RNP factory in the nucleoplasm (Tkacz et al., Reference Tkacz, Lustig, Stern, Biton, Salmon-Divon, Das, Bellofatto and Michaeli2007). When labelling active transcription through BrdU incorporation, a broader distribution of extra-nucleolar transcriptional activity can be observed apart from the SL-RNA arrays (although that accounts for Pol-III as well) (Daniels et al., Reference Daniels, Gull and Wickstead2010; illustrated in Fig. 2B). One would expect that capping enzymes and cap methyltransferases would concentrate at SL-RNP factories, which is difficult to extrapolate from the localization data currently available and will therefore require a more detailed analysis, possibly with higher resolution microscopy.
The VSG expression factory
African trypanosomes and their VSGs are a fine example of extreme biology and have led to several groundbreaking discoveries, such as trans-splicing, mRNA transcription by Pol-I or GPI anchors (Navarro et al., Reference Navarro, Peñate and Landeira2007; Duraisingh and Horn, Reference Duraisingh and Horn2016). Notably, recent studies on VSG expression in T. brucei have revealed interesting features regarding genome architecture and nuclear compartmentalization that hint to unknown layers of gene expression control in these organisms.
The single active VSG gene generates the most abundant protein in the cell (approximately 10% of the total proteome), which results from a combination of high levels of transcription by Pol-I and multiple mechanisms of post-transcriptional control (Navarro and Gull, Reference Navarro and Gull2001; Günzl et al., Reference Günzl, Bruderer, Laufer, Schimanski, Tu, Chung, Lee and Lee2003; do Nascimento et al., Reference do Nascimento, Egler, Arnold, Papavisiliou, Clayton and Erben2020; Viegas et al., Reference Viegas, de Macedo, De Niz, Rodrigues, Aresta-Branco, Jaffrey and Figueiredo2020). This renders trypanosomes and their VSGs an amenable model system to study mechanisms underpinning single gene choice, which are not fully understood in any eukaryote. Indeed, monogenic expression is one of the greatest outstanding mysteries of eukaryotic gene expression. For instance, it also underpins singular expression of antigen and olfactory receptors, responsible for the specificity of the immune response and the sense of smell in mammals, respectively (Monahan and Lomvardas, Reference Monahan and Lomvardas2015; Outters et al., Reference Outters, Jaeger, Zaarour and Ferrier2015).
Interestingly, it was unclear whether genome architecture and specifically genome position played a role in gene expression control in trypanosomes and related organisms. However, T. brucei somehow employs a mechanism of monogenic antigen transcription in the absence of controlled transcription initiation and canonical enhancer sequences. Indeed, Pol-I transcription is initiated at the same rate at all VSG-ESs, however transcription elongation is restricted to the active-VSG-ES (Vanhamme et al., Reference Vanhamme, Poelvoorde, Pays, Tebabi, Van Xong and Pays2000; Kassem et al., Reference Kassem, Pays and Vanhamme2014). Additionally, RNA maturation seems to be somehow restricted to the active VSG-ES suggesting that access to RNA processing factors or substrates might be limiting (Vanhamme et al., Reference Vanhamme, Poelvoorde, Pays, Tebabi, Van Xong and Pays2000; Kassem et al., Reference Kassem, Pays and Vanhamme2014).
Notably, while the silent VSG-ESs were located at more peripheral locations (Chaves et al., Reference Chaves, Zomerdijk, Dirks-Mulder, Dirks, Raap and Borst1998; Landeira and Navarro, Reference Landeira and Navarro2007), the active VSG-ES was included within an extra-nucleolar structure (although in close proximity to the nucleolus), designated the expression-site body (ESB), a transcription factory that contains a local reservoir of Pol-I (Navarro and Gull, Reference Navarro and Gull2001) (Fig. 3). This exclusion from the nucleolus is independent of the promoter, as swapping the VSG promoter by an rRNA promoter did not lead to nucleolar incorporation (Chaves et al., Reference Chaves, Zomerdijk, Dirks-Mulder, Dirks, Raap and Borst1998), suggesting that other DNA elements/factors are required for targeting.
The ESB emerged as the defining structure that sustained VSG monogenic expression, accommodating a single VSG-ES at a time. In fact, if two VSGs were simultaneously active, a dynamic colocalization with the ESB was observed (Chaves et al., Reference Chaves, Rudenko, Dirks-Mulder, Cross and Borst1999; Budzak et al., Reference Budzak, Kerry, Aristodemou, Hall, Witmer, Kushwaha, Davies, Povelones, McDonald, Sur, Myler and Rudenko2019). However, the mechanisms for targeting the active-VSG ES to the ESB as well as the protein composition and the exact DNA sequences incorporated within this structure have remained elusive. Although the complete molecular understanding is yet to be achieved, several major advances have taken place in the recent years.
Notably, the single active-VSG displays a specific inter-chromosomal interaction with a major mRNA splicing locus, one of the SL-RNA arrays, and this specific nuclear arrangement is critical to sustain VSG monogenic expression (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). Specifically, the single active-VSG is expressed within a dedicated sub-nuclear compartment harbouring the Pol-I transcribed antigen-coding gene and the Pol-II transcribed SL-array and their respective associated factors to ensure (1) monogenic antigen transcription and (2) efficient mRNA splicing (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020) (Fig. 3). The VSG exclusion proteins 1 and 2 (VEX1 and VEX2), which form discrete protein condensates in the nucleus in BSFs specifically, associate with the SL-RNA array and the active-VSG ES, respectively (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020) (Fig. 3). VEX1 was identified through a genetic screening (Glover et al., Reference Glover, Hutchinson, Alsford and Horn2016) and VEX2 through VEX1 affinity purification (Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019). From the two proteins, VEX2, an RNA-helicase, has the most critical role on VSG monogenic expression: following its depletion, the ESB collapses and trypanosomes simultaneously transcribe all VSG-ESs, subsequently exposing multiple VSGs on their surface (Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019). Further, following VEX2 knockdown, all VSG-ESs can access the SL-RNA arrays, showing that VEX2 somehow sustains this dedicated sub-nuclear compartment and an exclusive association between the single active-VSG and the SL-array (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). Additionally, besides maintaining an exclusive interaction between the active-VSG and the SL-array, VEX2 appears to fine-tune gene expression at the active-VSG locus (Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019, Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). It is tempting to speculate that it orchestrates a specific chromatin configuration that maximizes the interaction between the VSG gene itself (not the promoter or the ES-associated genes) and the SL-array.
Phase separation and transcriptional control
More recently, liquid–liquid phase separation (LLPS) has been proposed (opinion piece by Hnisz et al., Reference Hnisz, Shrinivas, Young, Chakraborty and Sharp2017) and later demonstrated (Guo et al., Reference Guo, Manteiga, Henninger, Sabari, Dall'Agnese, Hannett, Spille, Afeyan, Zamudio, Shrinivas, Abraham, Boija, Decker, Rimel, Fant, Lee, Cisse, Sharp, Taatjes and Young2019) to be a major regulatory mechanism for enhancer-mediated transcriptional control in mammalian cells. Enhancers are short (50–1500 bp) DNA regulatory elements that activate the transcription of specific genes to a much higher level than would be the case in their absence; they function as a platform for the recruitment of activators, transcription factors and the RNA polymerase components. These DNA elements have a distal location and are brought in proximity to the target gene through chromatin loops. Notably, nucleation of phase-separated multi-molecular assemblies at enhancer sequences can explain the formation of super-enhancers (clusters of enhancers; sometimes hundreds), their high sensitivity to transcription inhibition, enhancer-mediated patterns of transcriptional bursts and simultaneous activation of multiple genes by the same enhancer (Hnisz et al., Reference Hnisz, Shrinivas, Young, Chakraborty and Sharp2017). Notably, computational simulations have shown that LLPS can explain experimental observations that traditional models for transcriptional control cannot (Hnisz et al., Reference Hnisz, Shrinivas, Young, Chakraborty and Sharp2017).
Enhancer sequences have never been found in trypanosomes and related parasites, and given their polycistronic transcription and overall lack of controlled transcription initiation, such mechanisms were thought to be unlikely to operate. But is this really the case? Indeed, it was unclear whether and how genome architecture and genome position played a role in gene expression in these parasites. Could it be that trypanosomes evolved unconventional enhancers? This will be addressed in the ‘Discussion’ section.
Discussion
Despite the many open questions, previous studies following the depletion of several chromatin-associated factors (reviewed by Cestari and Stuart, Reference Cestari and Stuart2018) and the recently unveiled association between the active-VSG and the SL-array unequivocally demonstrate that genome architecture does play a role in VSG monogenic transcription in T. brucei. Further, spatial proximity to RNA-processing centres might be a conserved mechanism for post-transcriptional enhancement of gene expression but this had not previously been linked to inter-chromosomal interactions.
It is possible that all VSG-ESs are able to stochastically interact with the SL-arrays and compete for a limited pool of VEX2, which will then stabilize an exclusive interaction between a single VSG locus and the SL-array. This would render VEX2 a limiting factor, which is supported by its low abundance and tight regulation (Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019). Interestingly, this could be explained by an LLPS model (Hnisz et al., Reference Hnisz, Shrinivas, Young, Chakraborty and Sharp2017; Guo et al., Reference Guo, Manteiga, Henninger, Sabari, Dall'Agnese, Hannett, Spille, Afeyan, Zamudio, Shrinivas, Abraham, Boija, Decker, Rimel, Fant, Lee, Cisse, Sharp, Taatjes and Young2019); indeed, phase-separating proteins were shown to be capable of generating stable sub-nuclear structures from dynamic interactions in mammals (Shin et al., Reference Shin, Chang, Lee, Berry, Sanders, Ronceray, Wingreen, Haataja and Brangwynne2018). Multiple studies have shown that high local concentrations of specific proteins and nucleic acids (where RNAs appear to be major players) and cooperative interactions among these molecules are implicated in the formation of phase-separated bodies (Shin et al., Reference Shin, Chang, Lee, Berry, Sanders, Ronceray, Wingreen, Haataja and Brangwynne2018; Guo et al., Reference Guo, Manteiga, Henninger, Sabari, Dall'Agnese, Hannett, Spille, Afeyan, Zamudio, Shrinivas, Abraham, Boija, Decker, Rimel, Fant, Lee, Cisse, Sharp, Taatjes and Young2019). Recently, a family of RNA helicases has been identified as major regulators of the assembly of sub-nuclear compartments through LLPS (Hondele et al., Reference Hondele, Sachdev, Heinrich, Wang, Vallotton, Fontoura and Weis2019); therefore, it is tempting to speculate this might be the case of VEX2. In fact, specific post-translational modifications can trigger nucleation of phase-separated bodies; curiously, the active-ES resides within a hot spot of highly SUMOylated proteins (López-Farfán et al., Reference López-Farfán, Bart, Rojas-Barros and Navarro2014). Notably, the global role, if any, of LLPS and phase-separating proteins on genome organization in Trypanosomatids is yet to be investigated.
Inter-chromosomal interactions were thought to have a stochastic nature, indeed the existence of stable inter-chromosomal interactions has been a subject of debate as they were thought to be difficult to re-establish following cell division, possibly relying on error-prone mechanisms (Finn and Misteli, Reference Finn and Misteli2019). Consequently, their role on gene expression was rather dubious. The only other known stable interaction occurs in a terminally differentiated cell, and very interestingly, in another system subject to allelic exclusion. Indeed, olfactory neurons possess a multi-chromosomal super-enhancer that associates with the single active olfactory receptor gene (Monahan et al., Reference Monahan, Horta and Lomvardas2019). In trypanosomes, the association of the active-VSG with the SL-array appears reminiscent, but classic transcriptional enhancement was replaced by what appears to be post-transcriptional enhancement instead. Despite the attractive theoretical reasons for the presence of such an enhancer in malaria-causing parasites, Hi-C analysis was unable to identify such an element in the P. falciparum genome (Lemieux et al., Reference Lemieux, Kyes, Otto, Feller, Eastman, Pinches, Berriman, Su and Newbold2013).
In trypanosomes, proximity to the SL-array is likely to provide post-transcriptional enhancement due to a high local concentration of SL-RNA. A substantial amount of SL-RNA is therefore hijacked, so that RNA processing can keep pace with the high rate of transcription provided by Pol-I (Fig. 3). Notably, it will be interesting to identify other active VSG-ES-associated factors that take part in this antigen expression factory: it is entirely conceivable that a number of splicing factors and enzymes involved in polyadenylation might be concentrated in this compartment. This certainly adds a layer of post-transcriptional control that had not been previously characterized. Moreover, this association is also reminiscent of those between highly transcribed chromosome regions and NSs in mammals (Quinodoz et al., Reference Quinodoz, Ollikainen, Tabak, Palla, Schmidt, Detmar, Lai, Shishkin, Bhat, Takei, Trinh, Aznauryan, Russell, Cheng, Jovanovic, Chow, Cai, McDonel, Garber and Guttman2018; Kim et al., Reference Kim, Venkata, Hernandez Gonzalez, Khanna and Belmont2020). Whether the high transcription rate is the cause or a consequence of such association remains debatable. Similarly in T. brucei, whether the association with the SL-RNA array precedes the activation of the VSG locus, or whether it occurs afterwards merely providing post-transcriptional enhancement, remains unclear. Notably, in other organisms, co-transcriptional RNA processing can affect transcription elongation rates (Kornblihtt et al., Reference Kornblihtt, de la Mata, Fededa, Munoz and Nogues2004). How a specific VSG gene is activated over the other possible alleles remains a mystery, and those early events underpinning the establishment of an active transcriptional state are incredibly difficult to capture. In Plasmodium, for instance, antisense long-non-coding-RNAs play a key role in regulating var gene activation and mutually exclusive expression (Amit-Avraham et al., Reference Amit-Avraham, Pozner, Eshar, Fastman, Kolevzon, Yavin and Dzikowski2015).
Certainly several mechanisms simultaneously operate to constrain the inactive VSG-ESs and prevent their derepression. For instance, heterochromatin-based silencing in trypanosomes involves, among others, ISWI, RAP1 and histone deacetylase (DAC) 3 (Hughes et al., Reference Hughes, Wand, Foulston, Young, Harley, Terry, Ersfeld and Rudenko2007; Yang et al., Reference Yang, Figueiredo, Espinal, Okubo and Li2009; Wang et al., Reference Wang, Kawahara and Horn2010; reviewed by Duraisingh and Horn, Reference Duraisingh and Horn2016; reviewed by Cestari and Stuart, Reference Cestari and Stuart2018) (Fig. 3). The histone tri-methyltransferase DOT1B that targets H3K76, for instance, is required for rapid VSG-ES silencing and for an efficient transition from an active to a silent state (Figueiredo et al., Reference Figueiredo, Janzen and Cross2008). Also, both the integrity of the NL and histone H1 are critical to maintain condensed chromatin in silenced regions (DuBois et al., Reference DuBois, Alsford, Holden, Buisson, Swiderski, Bart, Ratushny, Wan, Bastin, Barry, Navarro, Horn, Aitchison, Rout and Field2012; Povelones et al., Reference Povelones, Gluenz, Dembek, Gull and Rudenko2012). Strikingly, T. brucei lacks H3K9me3, a well-characterized marker for heterochromatin, and HP1 (Berriman et al., Reference Berriman2005), which plays a key role in var gene silencing in Plasmodium (Brancucci et al., Reference Brancucci, Bertschi, Zhu, Niederwieser, Chin, Wampfler, Freymond, Rottmann, Felger, Bozdech and Voss2014). Further, in Plasmodium, the histone methyltransferase SET10 colocalizes with the active var gene (Volz et al., Reference Volz, Bártfai, Petter, Langer, Josling, Tsuboi, Schwach, Baum, Rayner, Stunnenberg, Duffy and Cowman2012) and NAD(+)-dependent histone deacetylases, Sir2A and Sir2B, are required for silencing of different var gene subsets (Tonkin et al., Reference Tonkin, Carret, Duraisingh, Voss, Ralph, Hommel, Duffy, Silva, Scherf, Ivens, Speed, Beeson and Cowman2009), but these histone modifiers do not appear to affect VSG silencing (Alsford et al., Reference Alsford, Kawahara, Isamah and Horn2007). Indeed, in both trypanosomes and malaria-causing parasites, repressive heterochromatin plays a critical role in silencing all but one antigen-coding gene for successful antigenic variation. However, different chromatin remodellers, histone readers/erasers and histone chaperones appear to be involved in this process in trypanosomes and Plasmodium (reviewed by Duraisingh and Horn, Reference Duraisingh and Horn2016).
In a broader perspective, post-transcriptional enhancement of gene expression through spatial proximity to RNA-processing centres might be particularly relevant in less complex eukaryotes, where canonical transcriptional enhancers have not been identified, and particularly in Trypanosomatids, where transcriptional regulation is limited. Nonetheless, it remains to be investigated whether this type of regulation extends beyond VSGs in T. brucei, and whether it plays a broader role in gene expression control in kinetoplastids.
Notably, in T. brucei, Hi-C and ChIP-Seq analyses revealed that other highly transcribed loci (e.g. tandem arrays that encode for histones, tubulin, heat shock proteins, etc.) can interact with the SL-RNA array in the mammalian-infective stage (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). Moreover, in insect-stage T. brucei, procyclin coding loci also interact with the SL-RNA array (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). Given the fact that Hi-C is a very sensitive technique, it can capture strong and stable but also stochastic and transient interactions (Finn and Misteli, Reference Finn and Misteli2019); therefore, it will be interesting to investigate whether the interactions above are stochastic or whether they are associated with stable and heritable structures at the single-cell level. In other words, are there any other transcription/splicing factories in T. brucei and possibly in other related parasites? Could the SL-array act as an unconventional and post-transcriptional enhancer? The fact that the tubulin gene loci in T. cruzi do not colocalize with the SL-RNA arrays (Dossin and Schenkman, Reference Dossin Fde and Schenkman2005) does not completely rule out this idea. This is not inconsistent with such interactions being transient and therefore more difficult to capture my microscopy, but could also mean that strong and stable interactions might be restricted to specific gene families and specific developmental stages, possibly depending on transcriptional activity. Additionally, it is very interesting that in T. brucei, the two SL-arrays occupy distinct chromosome territories, essentially there are two SL-RNA expression factories: is it because one is permanently used to sustain the expression of the active-VSG gene?
In mammals, a high degree of heterogeneity in genome organization has been observed, suggesting that individual cells in a population can assume many distinct, albeit related, spatial conformations (Finn et al., Reference Finn, Pegoraro, Brandão, Valton, Oomen, Dekker, Mirny and Misteli2019). Notably, such variability does not mean that chromatin organization has no functional relevance, but rather suggests that structural heterogeneity may be another layer impacting gene expression (Finn and Misteli, Reference Finn and Misteli2019). In Plasmodium for instance, the 3D genome structure appears to be strongly connected with the transcriptional activity of specific gene families throughout the life cycle (Bunnik et al., Reference Bunnik, Cook, Varoquaux, Batugedara, Prudhomme, Cort, Shi, Andolina, Ross, Brady, Fidock, Nosten, Tewari, Sinnis, Ay, Vert, Noble and Le Roch2018). Whether such variability can be observed in Trypanosomatid parasites and how that might modulate gene expression at the single-cell level and in different developmental stages remains to be unravelled.
Future directions
Trypanosoma brucei genome sequencing was a phenomenal turning point that marked the beginning of a new era of research. Since then, sequencing technology, gene-editing tools, imaging and affinity purification techniques have massively evolved, allowing us to experimentally tackle long-standing questions that had been previously untrackable.
Similarly to many pathogens, in T. brucei, the highly repetitive nature and heterozygosity of the antigen-gene arrays had precluded a complete genome assembly. Recently, through a combination of PacBio single-molecule real-time sequencing technology and Hi-C, the haplotype-specific assembly and scaffolding of the long antigen-gene arrays has been successful (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). This refined genome assembly has been proven critical to perform further analyses on chromatin organization and gene expression, especially regarding VSG genes. Among several downstream analyses, which largely benefitted from a refined genome assembly, is Hi-C.
Hi-C and other chromosome conformational capture techniques are a set of powerful molecular biology methods based on proximity labelling, which enable the analysis of chromatin spatial organization. These methods quantify the interaction frequency between genomic loci that are nearby in the 3D nuclear space, but may be far in the linear genome, allowing the identification of enhancer–promoter contacts or chromatin loops for instance (reviewed by Kempfer and Pombo, Reference Kempfer and Pombo2020). Hi-C studies in T. brucei have identified key architectural proteins and that a specific chromatin configuration is critical to fine-tune recombination events; indeed, perturbation of that specific architecture triggers switches in antigen expression (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). Further, virtual 4C analyses survey the interaction frequencies between a bait locus of interest and any other loci in the genome. In T. brucei, such analyses have demonstrated that the active-VSG ES (but not the silent) as well as genes encoding for other highly abundant proteins interact with the SL-array, uncovering a potential enhancer-like mechanism (Faria et al., Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020).
Next-generation sequencing techniques including RNA-Seq (transcript abundance), ChIP-Seq (chromatin-association) and CLIP-Seq (RNA-binding) have now been amply used in trypanosomes and other Trypanosomatids. More recently, ATAC-Seq (chromatin accessibility) and single-cell RNA-Seq have been performed in T. brucei (Muller et al., Reference Muller, Cosentino, Forstner, Guizetti, Wedel, Kaplan, Janzen, Arampatzi, Vogel, Steinbiss, Otto, Saliba, Sebra and Siegel2018). The latter opens unprecedented opportunities to investigate differential gene expression during developmental transitions and inherent single-cell variability within a particular life cycle stage.
Huge improvements have been made to imaging techniques and the fast pace of development is truly remarkable. In T. brucei, protein and DNA loci have been recently tracked at high resolution, using confocal-based or structured illumination microscopy (XY resolution 100–120 nm), which has been critical to characterize specific sub-nuclear compartments (Glover et al., Reference Glover, Hutchinson, Alsford and Horn2016; Budzak et al., Reference Budzak, Kerry, Aristodemou, Hall, Witmer, Kushwaha, Davies, Povelones, McDonald, Sur, Myler and Rudenko2019; Faria et al., Reference Faria, Glover, Hutchinson, Boehm, Field and Horn2019, Reference Faria, Luzak, Muller, Brink, Hutchinson, Glover, Horn and Siegel2020). But there is a growing need for methods that can image chromosomes with greater genomic and optical resolution; super resolution microscopy can now allow an XY resolution as low as 20–30 nm. To understand how the genome functions and regulates several key biological processes, it is necessary to visualize many genomic regions simultaneously, not just a few. Recently, there have been huge breakthroughs in other systems, such as OligoFISSEQ, a combination of three methods that employ fluorescence in situ sequencing (FISSEQ) of barcoded Oligopaint probes to enable the rapid visualization of multiple targeted genomic regions (Nguyen et al., Reference Nguyen, Chattoraj, Castillo, Nguyen, Nir, Lioutas, Hershberg, Martins, Reginato, Hannan, Beliveau, Church, Daugharthy, Marti-Renom and Wu2020). Another powerful technique is electron cryotomography, an imaging technique used to produce high-resolution 3D views of samples, typically biological macromolecules and cells. In trypanosomatids, it has been used to study flagellar and mitochondrial structures but to my knowledge, not to study supramolecular sub-nuclear complexes. For instance in humans, it was extensively used to study the human NPC (reviewed by Lin and Hoelz, Reference Lin and Hoelz2019).
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (Cas) technology has revolutionized molecular biology; indeed, it is a powerful tool that allows highly efficient and reproducible manipulation of genomic sequences for both locus-specific and genome-wide approaches. But its huge potential is not exclusively linked to the site-directed nuclease activity. A catalytically inactive Cas9 (dCas9) can be used as a universal recruitment platform in order to control transcription, visualize DNA sequences or investigate in situ proteomes (Anton et al., Reference Anton, Karg and Bultmann2018; Martens et al., Reference Martens, van Beljouw, van der Els, Vink, Baas, Vogelaar, Brouns, van Baarlen, Kleerebezem and Hohlbein2019). Indeed, for the identification of locus-associated proteins, dCas9 can be fused to a FLAG-tag and targeted to a locus of interest; chromatin is then crosslinked and fragmented; dCas9-bound chromatin fragments are subsequently isolated by FLAG-specific antibodies and analysed via mass spectrometry (enChIP) (Anton et al., Reference Anton, Karg and Bultmann2018). Unlike enChIP, CasID requires the expression of dCas9 fused to the promiscuous biotin ligase BirA*. After the culture medium has been supplemented with exogenous biotin, BirA* catalyses the addition of biotin to lysine residues of proteins that are in close proximity to the dCas9-BirA* fusion protein. Lysis of the cells and denaturation of proteins is then followed by affinity purification of biotinylated peptides, which are identified via tandem mass spectrometry (Anton et al., Reference Anton, Karg and Bultmann2018; Trinkle-Mulcahy, Reference Trinkle-Mulcahy2019). Indeed, this DNA-centric system can be used to pull-down proteins that associate with a specific locus; taking the VSG-ESs as an example, this system could help identifying factors specifically associated with the active or silent-ESs and factors involved in gene activation or gene silencing.
Additionally, several different dCas9-based systems have been developed to perform programmable control of spatial genome organization, among those is the CRISPR-genome organization (CRISPR-GO) system. It delivers a highly efficient and versatile control over the spatial positioning of genomic loci relative to specific nuclear compartments, including the nuclear periphery, CBs and promyelocytic leukaemia bodies to study how nuclear structure affects gene regulation and cellular function (Wang et al., Reference Wang, Xu, Nguyen, Liu, Gao, Lin, Daley, Kipniss, La Russa and Qi2018). For example, in T. brucei, this could be used to bring genomic loci in proximity to the SL-array or the NL and assess how that impacts gene expression. Recently, a CasDrop system was designed to study the formation of phase-separated compartments in the nucleus by enabling liquid condensation of transcriptional regulators at target loci (Shin et al., Reference Shin, Chang, Lee, Berry, Sanders, Ronceray, Wingreen, Haataja and Brangwynne2018). For example, in T. brucei, this could be used to investigate the formation of VEX2 protein condensates at the active VSG-ES. CRISPR/Cas9 technology has been successfully adapted to trypanosomes (reviewed by Bryant et al., Reference Bryant, Baumgarten, Glover, Hutchinson and Rachidi2019) and proven highly versatile; it will be interesting to see the future developments.
In summary, huge technological advances have been accomplished in the recent years and certainly many more will in the near future. This burst of technological breakthroughs will hopefully pave the way for future discoveries on nuclear and genome organization as well as gene expression control in African trypanosomes and related organisms.
Concluding remarks
Trypanosoma brucei nuclear organization and gene expression present several striking differences when compared to more complex eukaryotes. Multiple lines of evidence strongly support that its monogenic antigen transcription, which is critical for successful antigenic variation, is enforced and facilitated by a key nuclear architecture that involves specific inter-chromosomal interactions and compartmentalization (possibly also modification) of specific factors.
The molecular understanding of the mechanisms underpinning gene expression control in different developmental stages of these parasites is of great importance, as it might aid future vaccine and drug development efforts. For instance, acoziborole, a single-dose oral drug to treat trypanosomiasis, was shown to target cleavage and polyadenylation specificity factor 3 (Wall et al., Reference Wall, Rico, Lukac, Zuccotto, Elg, Gilbert, Freund, Alley, Field, Wyllie and Horn2018). Therefore, RNA processing is now established as a clinically validated drug target in the African trypanosome. Understanding the context within which drugs work can greatly facilitate the drug discovery process.
Notably, recent technological advances on sequencing, imaging and affinity purification techniques have led to important discoveries and paved the way to novel research avenues regarding nuclear organization and gene expression control in trypanosomes. Indeed, we live in exciting times where the pace of technology development is phenomenal and hopefully will allow us to address long-standing questions in infection biology that were previously inaccessible.
Acknowledgements
I would like to thank David Horn for the many exciting discussions about this subject and I would like to apologize to those whose work was not cited because of content and length constraints.
Financial support
J.R.C.F. is a senior research associate funded by a Wellcome Trust Investigator Award to Professor David Horn (100320/Z/12/Z).
Conflict of interest
None.
Ethical standards
Not applicable.




 Open access
Open access