



Introduction
Until recently, trypanosomatids parasitizing insects were a relatively poorly known group of flagellates with various transmission routes (Frolov et al., Reference Frolov, Kostygov and Yurchenko2021), particularly when compared to the well-studied members of the genera Trypanosoma and Leishmania, the causative agents of serious diseases of humans and other vertebrates (Maslov et al., Reference Maslov, Votýpka, Yurchenko and Lukeš2013). However, this has now changed as trypanosomatids are turning into model organisms suitable for addressing general questions in evolutionary, cell and molecular biology (Lukeš et al., Reference Lukeš, Speijer, Zíková, Alfonzo, Hashimi and Field2023). Many species can quickly achieve high-cell density in inexpensive media, are non-pathogenic for humans, and their assembled and annotated genomes became available (Albanaz et al., Reference Albanaz, Carrington, Frolov, Ganyukova, Gerasimov, Kostygov, Lukeš, Malysheva, Volf, Votýpka, Zakharova, Záhonová, Zimmer, Yurchenko and Butenko2023). Moreover, they are generally amenable to genetic modifications using a battery of tools extensively used in forward and reverse genetics of Trypanosoma brucei and Leishmania species (Matthews, Reference Matthews2015).
Phylogenetic studies revealed that monoxenous (single-host) trypanosomatids of invertebrates, now classified into 22 genera (Kostygov et al., Reference Kostygov, Albanaz, Butenko, Gerasimov, Lukeš and Yurchenko2024), are the predecessors of dixenous (2-host) phytomonads, leishmanias, and trypanosomes (Lukeš et al., Reference Lukeš, Skalický, Týč, Votýpka and Yurchenko2014). Therefore, the former flagellates emerged as organisms important for our understanding of complex life cycles, pathogenicity, and the unusual cell and molecular features of the latter serious human pathogens. Furthermore, trypanosomatids belonging to the genera Angomonas, Strigomonas, Kentomonas, Novymonas, and Phytomonas contain symbiotic bacteria that appear to represent various stages in the gradual transition of the endosymbiont into an organelle fully controlled by the host (De Souza and Motta, Reference De Souza and Motta1999; Kostygov et al., Reference Kostygov, Butenko, Nenarokova, Tashyreva, Flegontov, Lukeš and Yurchenko2017; Ganyukova et al., Reference Ganyukova, Frolov, Malysheva, Spodareva, Yurchenko and Kostygov2020). Consequently, they are likely to become particularly informative for the studies of symbiosis (Husnik et al., Reference Husnik, Tashyreva, Boscaro, George, Lukeš and Keeling2021; Zakharova et al., Reference Zakharova, Tashyreva, Butenko, Morales, Saura, Svobodová, Poschmann, Nandipati, Zakharova, Noyvert, Gahura, Týč, Stuhler, Kostygov, Nowack, Lukeš and Yurchenko2023). Members of the morphologically conspicuous genus Blastocrithidia were shown to have a reassigned genetic code with all 3 stop codons coding for amino acids (Záhonová et al., Reference Záhonová, Kostygov, Ševčíková, Yurchenko and Eliáš2016). Even more importantly, a novel mechanism responsible for this departure from the canonical genetic code has been discovered in Blastocrithidia nonstop (Kachale et al., Reference Kachale, Pavlíková, Nenarokova, Roithová, Durante, Miletínová, Záhonová, Nenarokov, Votýpka, Horáková, Ross, Yurchenko, Beznosková, Paris, Valášek and Lukeš2023) and may have a wider occurrence (Baranov and Atkins, Reference Baranov and Atkins2023).
Another interesting aspect of trypanosomatid biology is their emerging extreme diversity. Early estimates based on the ‘one-host, one-parasite paradigm’ and the observed high prevalence in the dipteran and hemipteran insects (Podlipaev et al., Reference Podlipaev, Sturm, Fiala, Fernandes, Westenberger, Dollet, Campbell and Lukeš2004) led to the prediction of over a million species (Stevens, Reference Stevens2001). However, more in-depth studies showed, both experimentally and under natural conditions, that this paradigm does not hold and that the same trypanosomatid species can infect several host species across continents (Kostygov et al., Reference Kostygov, Grybchuk-Ieremenko, Malysheva, Frolov and Yurchenko2014; Králová et al., Reference Králová, Grybchuk-Ieremenko, Votýpka, Novotný, Kment, Lukeš, Yurchenko and Kostygov2019; Votýpka et al., Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a, Reference Votýpka, Kment, Yurchenko and Lukeš2020). Furthermore, insect orders other than Hemiptera, Diptera, and Siphonaptera are only rarely infected and their trypanosomatid fauna is much less speciose. Inevitably, these observations, further strengthened by the cosmopolitan presence of some trypanosomatids (e.g. B. nonstop and Crithidia mellificae) and their repeated encounters in different continents (Králová et al., Reference Králová, Grybchuk-Ieremenko, Votýpka, Novotný, Kment, Lukeš, Yurchenko and Kostygov2019; Votýpka et al., Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a, Reference Votýpka, Klepetková, Yurchenko, Horák, Lukeš and Maslov2012b, Reference Votýpka, Kment, Yurchenko and Lukeš2020; Dario et al., Reference Dario, Lisboa, Silva, Herrera, Rocha, Furtado, Moratelli, Roque and Jansen2021) imply that the number of extant trypanosomatid species is orders of magnitude lower.
Still, one aspect significantly complicates this picture, namely the observation that morphologically diverse flagellates are rather frequently encountered in a single insect host (Yurchenko et al., Reference Yurchenko, Kostygov, Havlová, Grybchuk-Ieremenko, Ševčíková, Lukeš, Ševčík and Votýpka2016; Frolov et al., Reference Frolov, Malysheva, Ganyukova, Yurchenko and Kostygov2017, Reference Frolov, Malysheva, Ganyukova, Spodareva, Yurchenko and Kostygov2019). This finding was further corroborated by the serendipitous observation that trypanosomatids established in culture quite often differ from those detected by sequencing in the original host (Kostygov et al., Reference Kostygov, Malysheva and Frolov2011, Reference Kostygov, Grybchuk-Ieremenko, Malysheva, Frolov and Yurchenko2014), providing additional evidence of more than a single parasite species per host. Consequently, it can be assumed that using the PCR-based detection, genes from the most abundant flagellate species are preferentially amplified, with the co-occurring parasites going unnoticed. Since co-infections with several Trypanosoma species are common in Glossina flies, mixed infections of monoxenous trypanosomatids would not be surprising.
Therefore, we aimed to disentangle the composition of mixed trypanosomatid infections. For this, we have used a set of samples isolated from the hemipteran and dipteran insects captured in Cuba. Nanopore sequencing technology allowed us to determine that indeed, in an individual host specimen, up to 8 trypanosomatid species may co-occur. Although not species-specific and limited to a particular ecological group, when reasonably extrapolated to tropical insect diversity, such co-infections could again substantially increase the plausible species richness and host specificity of trypanosomatids, concealed by their morphological uniformity.
Materials and methods
Fieldwork, cultivation and host identification
True bugs and flies have been collected by using sweep netting and handpicking in several Cuban locations (Cienfuegos: 22°10'N, 80°24'W, 20 m; Las Palmas: 22°43'N, 83°32'W, 145 m; Matanzas: 23°1'N, 81°29'W, 40 m; Palma Rubia: 22°51'N, 83°27'W, 5 m; Soroa: 22°46'N, 83°0'W, 80 m; Topes de Collantes: 21°53'N, 80°1'W, 650 m; Trinidad: 21°47'N, 79°58'W, 35 m; Vinales: 22°36'N, 83°44'W, 140 m). The insects were dissected within 12 h of capture and the infected tissues were processed for DNA, smears and cultivation in the brain heart infusion medium supplemented with antibiotics, following a protocol described elsewhere (Lukeš and Votýpka, Reference Lukeš, Votýpka, Michels, Ginger and Zilberstein2020). The digestive tract was gently removed in a way that did not compromise the insect except for a few abdominal segments and if possible, it was divided into midgut (mesenteron) and hindgut (proctodeum). Following the establishment of species identity of the wet or dry-mounted specimens, these have been deposited in the National Museum of the Czech Republic, Prague. Upon transport to the laboratory, an axenic culture was established from a subset of trypanosomatid-positive samples, sometimes taking up to 6 months of continuous cultivation at room temperature without shaking.
DNA extraction, amplification and sequencing
Total genomic DNA was isolated from gut tissues or, in the case of successful cultivations, from 1 mL of axenically grown cultures, using a protocol described previously (Votýpka et al., Reference Votýpka, Kostygov, Kraeva, Grybchuk-Ieremenko, Tesařová, Grybchuk, Lukeš and Yurchenko2014). To amplify the 18S rRNA trypanosomatid gene, ~10 ng of DNA was subjected to nested PCR following Seward et al. (Reference Seward, Votýpka, Kment, Lukeš and Kelly2017). Sequences of PCR products obtained by Sanger sequencing were checked using Geneious software (version 10.0.6, https://www.geneious.com) and if mixed infections occurred, Oxford Nanopore Technologies (ONT) sequencing was applied, with the same total genomic DNA and outer primers as used for PCR amplification. However, the conditions were optimized for use in a one-step PCR amplification. Briefly, the PCR reaction contained TrN-F2 and TrN-R2 primers at final concentration of 400 nM each, AccuTaq polymerase (1U/25 μL), PCR buffer (Sigma) supplemented with 1 M betain and 1% DMSO. The number of PCR cycles (n) ranged from 24 to 33 and has been optimized for individual samples to prevent overcycling. The PCR conditions were as follows: denaturation step 95°C for 2 min, amplification n-times (95°C for 15 s, 63°C for 30 s, and 65°C for 5 min), and final extension step at 68°C for 20 min. The extended elongation time has been important both for effective amplification and prevention of artefacts.
Subsequently, libraries for ONT sequencing were prepared from purified PCR products using a ligation sequencing (SQK-LSK109) and native barcoding expansion 1-12 and 13-24 (EXP-NBD104 and EXP-NBD114) kits, according to the manufacturer´s instructions. Libraries were sequenced on the ONT GridION platform using the R9.4 chemistry (Flow-Cell). Sequencing data were base-called, i.e. transmission from physical changes in the electric current signal measured by the ONT sequencing device to biologically relevant bases, using Guppy v.5.1.13 (Wick et al., Reference Wick, Judd and Holt2019).
The sequencing data were processed using the Porechop v.0.2.4 pipeline to trim the barcodes and to discard possible technical chimeric reads, i.e. reads with barcode or any other technical sequence in the middle of the read (Wick et al., Reference Wick, Judd, Gorrie and Holt2017). Then, we utilized the NanoCLUST pipeline to resolve the species clusters and/or representative sequences. All samples were processed using the following parameters: –min_read_length 1800 –max_read_length 2100 –cluster_sel_epsilon 1 and the limit for cluster size was set as 4% of the total reads for a given sample. When such parameters led a to crash of the pipeline in any step, we tweaked –cluster_sel_epsilon and minimal cluster size until the pipeline identified reasonable clustering within the data, based on visual inspection. In such a case, only the clusters/sequences that fulfilled the condition of cluster size = > 4% of total reads were reported. Finally, the proportional representation of the individual species clusters was estimated using a custom R script (R core team, 2020). When cultivation provided 18S rRNA sequences that were missing from the ONT sequencing, we searched for such sequences either by relaxing minimal cluster size of the NanoCLUST pipeline or by direct search of the original data using BLASTn (Camacho et al., Reference Camacho, Coulouris, Avagyan, Ma, Papadopoulos, Bealer and Madden2009).
Phylogeny
Alignments for phylogenetic analysis were generated by MAFFT v.7 using the related sequences available in GenBank of the (nearly) full-size 18S rRNA gene. The final dataset contains 2222 characters and 131 sequences representing formally described species or molecular operational taxonomic units (mOTUs), which constitute proxy (geno)species, represented in some cases by several genotypes. Phylogenetic reconstructions were performed using maximum likelihood (ML; PhyML v.3.0.1) and Bayesian inference (BI; MrBayes v.3.2.2) with model optimization in ModelTest v.3.0.6. A general time-reversible substitution model with a mixed model for the variation of site rate (GTR + Γ + I) was chosen as the best-fit model for sequence evolution. Bootstrap analyses involved heuristic searches with 1000 replicates (maximum likelihood). Bayesian inference analysis was run for 5 million generations with covarion and sampling every 100 generations. All other parameters were left in their default states.
Nomenclature
To prevent any confusion regarding the ‘Newbiana’ and ‘Muscomonas’ nomenclature, we herein disclaim these two informal names (given in quotation marks and not italicized) for nomenclatural purposes according to The International Code of Zoological Nomenclature, Chapter 3, Article 8.3. (ICZN 1999, https://www.iczn.org/the-code/the-code-online/), thus preventing them from becoming available and entering any possible homonymy until properly described. We recommend authors using the names in question to follow the same notation method.
Results
Trypanosomatids infecting hemipteran hosts
Out of 417 dissected hemipterans (all from the suborder Heteroptera) belonging to 16 families and 44 species, 74 specimens (18%) were detected by microscopic examination to be infected with trypanosomatids (Tables 1 and 2). Particularly high prevalence was observed for the families Pyrrhocoridae (28 examined/10 infected/36% prevalence), Lygaeidae (56/14/25%), Rhopalidae (132/31/23%), and Alydidae (10/2/20%), with the genera Niesthrea (Rhopalidae), Ochrostomus (Lygaeidae), and Dysdercus (Pyrrhocoridae) accounting for 2 thirds of all positive samples. In Largidae (2/2/100%) and Reduviidae (4/2/50%) the prevalence was even higher, however, only a low number of individuals from these families was dissected.
Table 1. Summarized information about Cuban true bug (Heteroptera) hosts and their trypanosomatids, including the prevalence of parasites (number of dissected vs infected specimens) and the list of detected trypanosomatid species based on Sanger and Nanopore sequencing following the (nested) PCR of the homogenized host intestine and cultivation

Table 2. Summarized information about the detected trypanosomatids in Heteroptera insect host species (including number of infected specimens), localization of the infection in the host intestine and availability in culture

* Localization in host: HG, hindgut; MG, midgut; less infected part is in brackets.
Based on nested PCR and subsequent Sanger sequencing, the presence of trypanosomatids has been confirmed in all microscopically infected specimens (Tables 1 and 2). Co-infection of two trypanosomatid species occurred in 10 cases (13.5%), of which 8 were confirmed using ONT sequencing. Most of the mixed infections were confined to the family Rhopalidae (7 cases), but this is likely due to the large number of dissected individuals. The most common combination of trypanosomatid parasites, documented in 4 cases, was that of members of the genera Phytomonas and Obscuromonas.
In total, 21 different trypanosomatid species (due to missing formal descriptions, we refer to them here as molecular operational taxonomic units [mOTUs]) were found in the examined hemipterans, of which 11 were detected for the first time. We label them as new species based on differences in the 18S rRNA sequences considered as significant for this group of flagellates. However, all detected mOTUs from the hemipteran hosts can be affiliated with already known taxa and thus do not represent any major novel lineages or genera (Figs 1–3). More than half of the new mOTUs belong to the subfamily Blastocrithidiinae, with 5 falling into the genus Blastocrithidia. While Blastocrithidia sp. 4 is closely related to (or possibly conspecific with) the isolate CH322 from China, the other 4 sequences clearly represent new species, substantially extending the known diversity of this genetically remarkable genus. Since none of the new species was found in more than 2 hemipterans (Table 2), information on their host specificity remains limited. On the other hand, several widely distributed generalist species, such as B. nonstop, the Blastocrithidia papi/largi species complex and Obscuromonas oborniki, were detected in several hemipteran families and hence possess a wide host range. Although Obscuromonas sp. TU73b was found in 7 bugs from the family Rhopalidae, it was invariably present in co-infections with other trypanosomatids, be it members of the genera Phytomonas, Leptomonas or other Obscuromonas species.

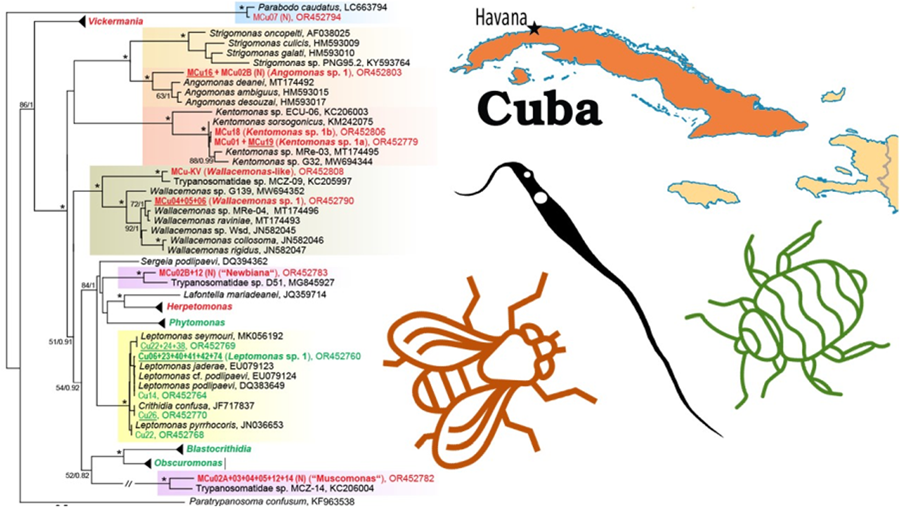
Figure 1. An 18S rRNA-based maximum likelihood phylogenetic reconstruction; sequences from Cuban heteropteran bugs (Cu) are indicated by green, from Cuban dipteran flies (MCu) by red, new species (mOTUs, molecular operational taxonomic units) by bold; isolates with culture established are underlined, (N) indicates detection only by Nanopore sequencing; for details of 5 selected genera see the individual subtrees (Figs 2–4, the number in brackets indicates the number of detected/number of new (in bold) mOTUs); asterisks mark branches with maximal statistical support (ML > 95, Bayesian > 0.95); double crossed branch is 50% of the original length; the scale bar denotes the number of substitutions per site.

Figure 2. Expanded subtree of the genus Blastocrithidia and Obscuromonas; for more detail see Fig. 1.

Figure 3. An 18S rRNA-based maximum likelihood phylogenetic reconstruction of the genus Phytomonas specifying the insect hosts (vectors); the blue undercolouring indicates species that are exclusively associated with the family Pentatomidae; for more detail see Fig. 1.
From the subfamily Herpetomonadinae, only the genus Phytomonas was present in the examined hemipterans (Figs 1 and 3). Four new mOTUs could be distinguished, although Phytomonas spp. 2 and 3, both from various shield bugs (Pentatomidae), might be considered conspecific, as their 18S rRNA genes differ in only 4 nucleotides. Phytomonas sp. 1 was repeatedly found in Niesthrea bugs (Rhopalidae) from several localities, making it the most encountered trypanosomatid in our survey. In one locality, this flagellate infected 2 individuals of Neomegalotomus rufipes (Alydidae), out of which one was coinfected with an already known Phytomonas sp. TU241 (Fig. 3, Table 2).
Of the 5 mOTUs detected within the subfamily Leishmaniinae, 4 could be assigned to already known species, with only 1 being novel (Fig. 1). Leptomonas sp. 1 was quite abundant and confined mostly to a single-host species, Ochrostomus pulchellus (Lygaeidae). While Leptomonas podlipaevi was detected only in one individual of Rhopalidae, Leptomonas pyrrhocoris, a cosmopolitan specialist associated with fire bugs (Pyrrhocoridae), as well as Leptomonas seymouri, were repeatedly found in several Dysdercus species. Finally, Crithidia confusa was encountered in a single co-infection with Blastocrithidia, being detected only in an established culture.
Trypanosomatids infecting dipteran hosts
Of 201 dissected fly specimens, only 20 (10%) were detected by microscopic examination to be infected with trypanosomatid parasites. Of these, nested PCR and subsequent Sanger sequencing revealed co-infection in 11 cases (55%), of which all were confirmed by ONT sequencing (Table S1).
Out of 20 microscopically infected flies, 10 and 5 were members of Muscidae and Sepsidae, respectively, while the remaining 5 infected flies belonged to the families Ulidiidae (2 flies), Calliphoridae, Drosophilidae and Lauxaniidae. Since uninfected flies were not taxonomically examined, the prevalence in individual families cannot be established.
Interestingly, 14 of the infected individuals (mostly belonging to Muscidae and Sepsidae) were inhabited by multiple trypanosomatid or bodonid species. In 11 cases, we were able to ascertain by deep ONT sequencing the identity of almost all trypanosomatids present in a single host, allowing us to document up to 8 species in one host specimen (MCu02 and MCu12) (Table S1, Figs 1, 3, 4 and S1). As a result, a total of 27 mOTUs were recognized, of which 25 can be considered as new species. Among them, 2 species seem to be so distant from their known relatives that their accommodation into new genera would be justifiable (see below). Trypanosomatids of the subfamily Herpetomonadinae were represented in our dataset by 5 mOTUs confined to Muscidae. Two of them, each found in one individual only, can be assigned to Herpetomonas samuelpessoai and H. modestus (Fig. 4A). While Herpetomonas sp. 2, found in 2 samples, is clearly a distinct species, the status of Herpetomonas spp. 1 and 3, present only in a single examined host, remains uncertain.

Figure 4. Expanded subtree of the genus Herpetomonas (A) and Vickermania (B); for more detail see Fig. 1.
The symbiont-containing subfamily Strigomonadinae comprises 2 new mOTUs. Since we succeeded in introducing both into the culture, they will be subject to a detailed examination in the future. Angomonas sp. 1 was present in 2 individuals of Calliphoridae and Muscidae and constitutes a basal lineage of the genus (Fig. 1). Two genotypes of Kentomonas sp. 1, related to K. sorsogonicus (Fig. 1), were identified in 2 Muscidae and one Ulidiidae flies, respectively. Two mOTUs fall into the genus Wallacemonas. Being a typical member of the genus, Wallacemonas sp. 1 was detected in 3 flies (2 individuals from Muscidae and 1 from Ulidiidae) (Table S1, Fig. 1). On the contrary, the isolate MCu-KV, which originated from Drosophilidae, is closely related to MCZ-09 from Lauxaniidae and either forms a basal lineage of the genus or even qualifies, together with MCZ-09, as a candidate for a new genus (Fig. 1).
The surprisingly high diversity of the genus Vickermania (sensu Kostygov et al., Reference Kostygov, Frolov, Malysheva, Ganyukova, Chistyakova, Tashyreva, Tesařová, Spodareva, Režnarová, Macedo, Butenko, D'avila-Levy, Lukeš and Yurchenko2020) revealed 16 mOTUs (Fig. 4B), all hitherto unknown. Most of them exhibit some level of host specificity, with Vickermania sp. 3 being confined to Calliphoridae, Vickermania sp. 4 to Lauxaniidae, Vickermania spp. 1, 7, and 10 to Sepsidae and, finally, Vickermania spp. 8, 9, 11 through 16 to Muscidae. Only Vickermania sp. 2 parasitized both Sepsidae and Ulidiidae, but even then, it was represented by 2 different genotypes, which could also be considered as 2 different mOTUs and therefore species. While the prevalence of members of the genus Vickermania was generally low, with only half of the mOTUs found in more than a single specimen, their co-infections were rather frequent in flies belonging to Muscidae and Sepsidae. As a result, 12 out of 16 mOTUs could only be detected by ONT sequencing, being masked in standard PCR by a more abundant flagellate (Table S1, Fig. 4B).
Moreover, application of this technology also allowed the detection of trypanosomatids that, based on the 18S rRNA sequences, represent 2 candidate new genera, both with unstable position in the phylogenetic tree. The first lineage, here tentatively named ‘Muscomonas’, was detected during this survey in 6 individuals of Muscidae, invariably in a co-infection with Vickermania spp. and/or another trypanosomatid. This candidate for a new genus is related to MCZ-14 from Opomyzidae captured in the Czech Republic (Fig. 1). The second group, here informally named ‘Newbiana’ (based on its provisional name ‘New-B’), was found in two individuals of Muscidae, with the most closely related sequence coming from a chimpanzee feacal sample from Cameroon (Fig. 1), although it has been proposed that the detected trypanosomatid originates from a fly that contaminated the sample (Votýpka et al., Reference Votýpka, Pafčo, Modrý, Mbohli, Tagg and Petrželková2018). In both cases, only sequences are available in the absence of any morphological data, because in the dry smears prepared from the infected insects (data not shown), it is technically challenging to associate a given cell with a given sequence.
Finally, in one Sepsidae (MCu-07), Parabodo caudatus was detected (Fig. 1), while another specimen from the same family (MCu-10) carried even 3 mOTUs belonging to the genus Parabodo (Table S1), representing likely a passive passage of free-living flagellates from water.
Trypanosomatid co-infections in dipteran hosts
The ONT sequencing enabled the identification of multiple trypanosomatid species co-infecting a single host, as well as the estimation of how numerous each species was. Furthermore, in some Muscidae, the midgut and the hindgut were dissected, microscopically examined and further processed separately, allowing to compare the occurrence and abundance of each trypanosomatid species in different parts of the digestive system.
The fly midgut was generally dominated by several Vickermania species or by the ‘Muscomonas’ flagellates (Table S1). An example of a heavy co-infection of several species is shown for MCu-12 (Fig. S1) and MCu-02 (Fig. 5A). Other trypanosomatids, including several Vickermania species, were much less numerous, indicating either their competitive exclusion by the dominant species, or an accidental or perennial midgut infection.

Figure 5. Both microscopically positive parts of the digestive tract of the muscid fly MCu02 were processed separately. By nanopore sequencing, 5 trypanosomatid species were detected in the midgut (left). While 2 Vickermania species (Vickermania sp. 8 and sp. 13) and new genus ‘Muscomonas’ are dominant inhabitants of the midgut, 2 Herpetomonas species (Herpetomonas sp. 1 and sp. 2) likely represent contaminants from the hindgut or passively passaged cells. In the hindgut (right), 8 trypanosomatids have been detected, of which Herpetomonas sp. 1 is the predominant species. Sequences derived from other species occur only in very low numbers and represent either cells released from the anterior part of the digestive tract (Vickermania sp. 8 and sp. 13 and ‘Muscomonas’) or cells that have been only passively passaged. Alternatively, they have been outcompeted by the dominant Herpetomonas species (in case of ‘Newbiana’, Herpetomonas sp. 2 and sp. 3, and Angomonas).
In the hindgut, members of the genera Herpetomonas or Kentomonas prevailed (e.g., MCu-02B and MCu-19), while other flagellates were rare (Fig. 5B). When both segments of the digestive tract of a dissected specimen were infected simultaneously, a small number of typical midgut inhabitants, such as ‘Muscomonas’ and Vickermania spp., also occurred in the hindgut (Fig. 5B). The same applies vice versa, as some hindgut dwelling trypanosomatids, such as members of the genus Herpetomonas, can be found in the midgut (Fig. 5A). However, when one segment of the digestive tract was infected, only the corresponding trypanosomatids were found (Fig. S1). For example, ‘Muscomonas’ and Vickermania spp. were never found in flies with only their hindgut infested, and similarly, Herpetomonas spp. were not detected in hosts with an infection confined to the midgut.
Discussion
While the last decade has seen a significant expansion of the known diversity of insect and plant trypanosomatids including some studies from Asia, Europe and Africa (Votýpka et al., Reference Votýpka, Maslov, Yurchenko, Jirků, Kment, Lun and Lukeš2010, Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a; Lukeš et al., Reference Lukeš, Butenko, Hashimi, Maslov, Votýpka and Yurchenko2018; Frolov et al., Reference Frolov, Malysheva, Ganyukova, Spodareva, Yurchenko and Kostygov2019, Reference Frolov, Malysheva, Ganyukova, Spodareva, Králová, Yurchenko and Kostygov2020), it was continental South and Central America where most sampling occurred (Teixeira et al., Reference Teixeira, Takata, Conchon, Campaner and Camargo1997, Reference Teixeira, Borghesan, Ferreira, Santos, Takata, Campaner, Nunes, Milder, De Souza and Camargo2011; Maslov et al., Reference Maslov, Westenberger, Xu, Campbell and Sturm2007, Reference Maslov, Yurchenko, Jirků and Lukeš2010; Borghesan et al., Reference Borghesan, Ferreira, Takata, Campaner, Borda, Paiva, Milder, Teixeira and Camargo2013; Kozminsky et al., Reference Kozminsky, Kraeva, Ishemgulova, Dobáková, Lukeš, Kment, Yurchenko, Votýpka and Maslov2015; Dario et al., Reference Dario, Lisboa, Silva, Herrera, Rocha, Furtado, Moratelli, Roque and Jansen2021). However, except for a single study conducted on the island of Curacao (Votýpka et al., Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019), so far, no information was available on these interesting protists in the Caribbean. Here, we present such a study from Cuba, in which we have not only mapped the distribution and diversity of trypanosomatids but also applied ONT sequencing to examine their frequency and extent of co-infections within a single insect host.
Trypanosomatid diversity in hemipteran hosts
Similar to other studies carried out elsewhere (Sbravate et al., Reference Sbravate, Campaner, Camargo, Conchon, Teixeira and Camargo1989; Votýpka et al., Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a, Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019, Reference Votýpka, Kment, Yurchenko and Lukeš2020; Kozminsky et al., Reference Kozminsky, Kraeva, Ishemgulova, Dobáková, Lukeš, Kment, Yurchenko, Votýpka and Maslov2015; Králová et al., Reference Králová, Grybchuk-Ieremenko, Votýpka, Novotný, Kment, Lukeš, Yurchenko and Kostygov2019), the highest prevalence of trypanosomatids was detected in Pyrrhocoridae, Lygaeidae, Rhopalidae, Alydidae, Pentatomidae, Reduviidae, and Largidae, with Rhyparochromidae, Scutelleridae, and Miridae being infected only rarely. While the lack of infections in Coreidae, Gerridae, and Nabidae can be explained by the low number of dissected individuals, the negativity of Oxycarenidae was unexpected, since the European population of Oxycarenus hyalinipennis and O. lavaterae harbour several trypanosomatid species (Franchini, Reference Franchini1922; Antonucci, Reference Antonucci1941; Seward et al., Reference Seward, Votýpka, Kment, Lukeš and Kelly2017). This may be caused by O. hyalinipennis being a non-native species introduced to America in 20th century (Grillo, Reference Grillo1993).
Based on the 18S rRNA sequences, 21 mOTUs could be distinguished in the studied bugs, 10 of which were already known from previous studies. This includes a cosmopolitan B. nonstop, known to have an extensively reassigned genetic code, which is able to parasitize bugs from 8 families (Králová et al., Reference Králová, Grybchuk-Ieremenko, Votýpka, Novotný, Kment, Lukeš, Yurchenko and Kostygov2019; Kachale et al., Reference Kachale, Pavlíková, Nenarokova, Roithová, Durante, Miletínová, Záhonová, Nenarokov, Votýpka, Horáková, Ross, Yurchenko, Beznosková, Paris, Valášek and Lukeš2023), now also including Rhopalidae (this study). The Blastocrithidia papi/largi species complex was retrieved from 3 different host families, one of which is the predatory assassin bug (Reduviidae), although due to the low intensity of infection, accidental transmission of the parasites from an infected prey cannot be excluded. Their close relatives Obscuromonas sp. 87JS (TU18) and Obscuromonas sp. Re35 (TU73) are, regardless of their geographic location, restricted to Miridae and mostly Rhopalidae, respectively (Westenberger et al., Reference Westenberger, Sturm, Yanega, Podlipaev, Zeledón, Campbell and Maslov2004; Maslov et al., Reference Maslov, Westenberger, Xu, Campbell and Sturm2007; Votýpka et al., Reference Votýpka, Kment, Yurchenko and Lukeš2020; this study). Not only is such a host preference unusual for Blastocrithidiinae, but it is also worth noting that the latter mOTU always occurred in co-infection with some other trypanosomatids, be it Phytomonas, Leptomonas, or other Obscuromonas species.
Leptomonas seymouri deserves particular attention, as it has been repeatedly detected in human cutaneous lesions caused by Leishmania spp. in India and neighbouring countries (Ghosh et al., Reference Ghosh, Banerjee, Sarkar, Datta and Chatterjee2012; Singh et al., Reference Singh, Chikara and Sundar2013), and thus cannot be considered as just a frequent contaminant of laboratory cultures (Kraeva et al., Reference Kraeva, Butenko, Hlaváčová, Kostygov, Myškova, Grybchuk, Leštinová, Votýpka, Volf, Opperdoes, Flegontov, Lukeš and Yurchenko2015). Although originally described from the cotton stainer bug Dysdercus suturellus (Wallace, Reference Wallace1977), ever since this trypanosomatid has been found neither in Dysdercus nor in any other Heteroptera, leading to uncertainty about the identity of its true host (Kraeva et al., Reference Kraeva, Butenko, Hlaváčová, Kostygov, Myškova, Grybchuk, Leštinová, Votýpka, Volf, Opperdoes, Flegontov, Lukeš and Yurchenko2015). On the other hand, experimental infections were much more successful in Dysdercus (Moraes et al., Reference Moraes, Freymuller, Camargo and Milder1994) than in sand flies (Phlebotomus spp.), the putative vector of L. seymouri in human lesions (Kraeva et al., Reference Kraeva, Butenko, Hlaváčová, Kostygov, Myškova, Grybchuk, Leštinová, Votýpka, Volf, Opperdoes, Flegontov, Lukeš and Yurchenko2015). However, our current finding puts this problem to rest, confirming that L. seymouri infects Dysdercus under natural conditions. Specifically, its current distribution in its insect host is confined to the Americas (Wallace, Reference Wallace1977; Votýpka et al., Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019; this study), whereas this flagellate was detected in Leishmania lesions in the Old World (Ghosh et al., Reference Ghosh, Banerjee, Sarkar, Datta and Chatterjee2012). Although based on old studies (Blacklock, Reference Blacklock1923), Dysdercus is able to bite humans, the co-transmission with Leishmania in this way is highly improbable, and the vector of L. seymouri in human lesions thus remains unknown.
Among the new mOTUs, Leptomonas sp. 1 stands out due to the high prevalence in its main host, Ochrostomus pulchellus. Its detection also in Niesthrea sidae can be explained by the feeding of both hosts on the Malvaceae plants (Baranowski and Slater, Reference Baranowski and Slater1975). It is noteworthy that the latter host species was fairly often infected by Phytomonas sp. 1, which was also found in 2 specimens of Neomegalotomus rufipes that feeds on Fabaceae (Froeschner, Reference Froeschner1942; Ventura et al., Reference Ventura, Montalvan and Panizzi2000a). Alternatively, N. rufipes could have obtained Phytomonas sp. 1 from feeding on a dead N. sidae, since necrophagy has been observed in this genus (Ventura et al., Reference Ventura, Silva and Panizzi2000b), likely resulting in non-specific infections by otherwise specialized trypanosomatids (Votýpka et al., Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019). Although found in only a few hosts, Phytomonas spp. 2, 3 and 4 belong to a clade that seems to be confined to various Pentatomidae (Fig. 3), while other Phytomonas spp. infect mainly bugs from the superfamily Coreoidea. Moreover, the inability of Phytomonas from a coreoid host to infect pentatomid bugs was recently experimentally demonstrated (Malysheva et al., Reference Malysheva, Ganyukova and Frolov2023). Still, no such specificity can be observed in the plant host, as Phytomonas species infecting different plant families often cluster together and vice versa (Zanetti et al., Reference Zanetti, Ferreira, Serrano, Takata, Campaner, Attias, De Souza, Teixeira and Camargo2016). Therefore, it appears that Phytomonas spp. are primarily specialized to the insect host, being confined to a single family or superfamily, whereas the spectrum of plant hosts can be much broader. Similar situation has been documented for the dixenous genus Leishmania. Indeed, some Leishmania species infect multiple various vertebrate host species, yet are restricted to a single insect species (Akhoundi et al., Reference Akhoundi, Kuhls, Cannet, Votýpka, Marty, Delaunay and Sereno2016).
Trypanosomatid diversity in dipteran hosts
Most infected flies belong to the families Muscidae and Sepsidae, which is likely due to their aggregative feeding on various liquids from dung. In total, we have identified 27 mOTUs from the dipteran hosts, of which only two were previously detected. Herpetomonas samuelpessoai has originally been described from an assassin bug Zelus leucogrammus (Galvão et al., Reference Galvão, Oliveira, Carvalho and Veiga1970), but since it was later encountered only in dipterans (Sarcophagidae, Anthomyiidae, and Muscidae) (Týč et al., Reference Týč, Votýpka, Klepetková, Suláková, Jirků and Lukeš2013; this study), it is likely that its true hosts are various brachycerans and the assassin bug infection was accidental. The other already known species is Herpetomonas modestus that has so far only been found in Muscidae (Týč et al., Reference Týč, Votýpka, Klepetková, Suláková, Jirků and Lukeš2013; this study) and Calliphoridae (Borghesan et al., Reference Borghesan, Ferreira, Takata, Campaner, Borda, Paiva, Milder, Teixeira and Camargo2013).
The trypanosomatid family Strigomonadinae invariably carries bacteria, and this symbiotic relationship is being studied in its South American isolates (Teixeira et al., Reference Teixeira, Borghesan, Ferreira, Santos, Takata, Campaner, Nunes, Milder, De Souza and Camargo2011; Borghesan et al., Reference Borghesan, Campaner, Matsumoto, Espinosa, Razafindranaivo, Paiva, Carranza, Añez, Neves, Teixeira and Camargo2018). Both new mOTUs also contain endosymbionts (data not shown), with one of them constituting the most basal lineage of the genus Angomonas. Same as other Angomonas species, it parasitizes the hindgut and midgut of Muscidae and Calliphoridae (Ganyukova et al., Reference Ganyukova, Malysheva and Frolov2017). The second novel mOTU (Kentomonas sp. 1) is closely related to K. sorsogonicus, yet instead of infecting Sarcophagidae (Votýpka et al., Reference Votýpka, Kostygov, Kraeva, Grybchuk-Ieremenko, Tesařová, Grybchuk, Lukeš and Yurchenko2014), this Cuban isolate was found in Muscidae and Ulidiidae.
The recently established genus Vickermania (Kostygov et al., Reference Kostygov, Frolov, Malysheva, Ganyukova, Chistyakova, Tashyreva, Tesařová, Spodareva, Režnarová, Macedo, Butenko, D'avila-Levy, Lukeš and Yurchenko2020) accommodated only 2 species so far, both isolated from Calliphoridae and Sepsidae. Hence, the discovery herein of numerous mOTUs infecting flies from Muscidae, Sepsidae, Calliphoridae, Ulidiidae, and Lauxaniidae is surprising and indicates either a particularly extensive diversity of this genus on the island or its hitherto overlooked presence elsewhere. Indeed, most Vickermania mOTUs have been detected only via ONT sequencing, which clearly demonstrates the power and utility of this approach (with the capability to sequence full-length target genes) and indicates that the latter possibility is the case, namely that these flagellates have often been overlooked in mixed infections. From the limited dataset available, rather narrow host specificity can be inferred, as no mOTU was detected in more than one dipteran family. Such a tight association is unusual among monoxenous trypanosomatids, since other Brachycera-infecting genera, such as Herpetomonas, Wallacemonas, and Crithidia, can be found in several dipteran families (Borghesan et al., Reference Borghesan, Ferreira, Takata, Campaner, Borda, Paiva, Milder, Teixeira and Camargo2013; Týč et al., Reference Týč, Votýpka, Klepetková, Suláková, Jirků and Lukeš2013). It is also surprising considering that Vickermania spadyakhi from Sepsidae was under experimental conditions able to infect Lucilia flies (Calliphoridae) (Kostygov et al., Reference Kostygov, Frolov, Malysheva, Ganyukova, Chistyakova, Tashyreva, Tesařová, Spodareva, Režnarová, Macedo, Butenko, D'avila-Levy, Lukeš and Yurchenko2020).
Among the detected flagellates, 2 groups, provisionally labelled ‘Muscomonas’ and ‘Newbiana’, deserve in terms of their sequence divergence the status of a new genera. However, the failure to introduce them into culture and describe their morphology precludes their formal description for the time being, as there is no feasible way to associate cells from mixed infections with a given sequence. One possible approach is single-cell sequencing; however, this would be most challenging with dry smears, where usually multiple cells are in close physical contact and the DNA is of poor quality. Both unnamed genera have been detected in the midgut and hindgut of Muscidae from several Cuban localities. Similarly, 2 previously published sequences clustering with these new mOTUs (Fig. 1) also originate from flies (MCZ-14) (Týč et al., Reference Týč, Votýpka, Klepetková, Suláková, Jirků and Lukeš2013) or are very likely derived from them (D51) (Votýpka et al., Reference Votýpka, Pafčo, Modrý, Mbohli, Tagg and Petrželková2018).
Co-infections, tissue localization and host specificity
The extremely high intraspecific and low interspecific morphological variability of trypanosomatids makes it almost impossible to distinguish in situ even distantly related species, genera or subfamilies (Podlipaev and Lobanov, Reference Podlipaev and Lobanov1996). For this reason, the infections of a single host by multiple species of trypanosomatids are very difficult to discern and were for over a century (Prowazek, Reference Prowazek1904) responsible for frequent confusions regarding species identity, morphology and life cycles. However, since the onset of the sequencing era, reliable species delimitations and identifications became possible, revealing, among other things, the commonality of co-infections (Votýpka et al., Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a, Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019; Lukeš et al., Reference Lukeš, Butenko, Hashimi, Maslov, Votýpka and Yurchenko2018; Králová et al., Reference Králová, Grybchuk-Ieremenko, Votýpka, Novotný, Kment, Lukeš, Yurchenko and Kostygov2019). Likely used for the first time in the study of insect trypanosomatids, ONT sequencing proved to be highly sensitive, revealing frequent co-infections constituted by abundant as well as (very) rare species. Furthermore, thanks to the relative ease of amplification and the comparable copy number of rRNA genes in different trypanosomatids (Albanaz et al., Reference Albanaz, Carrington, Frolov, Ganyukova, Gerasimov, Kostygov, Lukeš, Malysheva, Volf, Votýpka, Zakharova, Záhonová, Zimmer, Yurchenko and Butenko2023), by sequencing tens of thousands of reads, it is possible to estimate how many cells are there in a sample, thus indicating the strength of infection and identifying the (pre)dominant species. There are several other ways how to identify multiple trypanosomatid infection in insects, e.g. using PCR amplification of the spliced leader RNA gene (Kozminsky et al., Reference Kozminsky, Kraeva, Ishemgulova, Dobáková, Lukeš, Kment, Yurchenko, Votýpka and Maslov2015). However, in the terms of accuracy and estimation of relative quantification, ONT sequencing has obvious advantages over the PCR length-based approaches.
Within an insect host, different trypanosomatids inhabit different parts of its digestive and/or excretory tracts (Frolov et al., Reference Frolov, Kostygov and Yurchenko2021), resulting in a niche partitioning during co-infection. Such differentiation can be caused by distinct features of various life cycles. For example, in the tabanid Hybomitra solstitialis, Wallacemonas raviniae builds up massive loosely attached growths on the rectal wall, while Trypanosoma theileri adheres tightly to the ileum using an extracellular matrix. This likely reflects the transmission of the former species among its hosts through cells leaving the digestive tract, whereas the latter flagellate has no benefit in leaving the host, as its life cycle proceeds following the ingestion of an infected tabanid (Malysheva et al., Reference Malysheva, Frolov and Kostygov2022). In another case, in the herbivorous species of true bug Coreus marginatus, Phytomonas is transmitted to the host plant via infected salivary glands (Frolov et al., Reference Frolov, Malysheva, Ganyukova, Spodareva, Yurchenko and Kostygov2019), while the transmission of co-infecting Blastocrithidia among insect hosts occurs by a cyst-like stage, which is formed in the midgut and rectum (Frolov et al., Reference Frolov, Malysheva, Ganyukova, Spodareva, Králová, Yurchenko and Kostygov2020), resulting in functional niche partitioning. Indeed, the coinfections of Phytomonas and Obscuromonas or Blastocrithidia appears to be common among Coreidae, Rhopalidae, Alydidae and Pentatomidae (Votýpka et al., Reference Votýpka, Klepetková, Jirků, Kment and Lukeš2012a, Reference Votýpka, Kment, Kriegová, Vermeij, Keeling, Yurchenko and Lukeš2019; this study).
Moreover, a niche partition can be observed even within one segment of the digestive tract. The firebug Pyrrhocoris apterus is commonly co-infected by L. pyrrhocoris and B. papi, both inhabiting the midgut. While Leptomonas dwells exclusively in the lumen of the gut (Votýpka et al., Reference Votýpka, Klepetková, Yurchenko, Horák, Lukeš and Maslov2012b), Blastocrithidia usually attaches to the epithelium (Frolov et al., Reference Frolov, Malysheva, Ganyukova, Yurchenko and Kostygov2017). Furthermore, to multiply and produce cyst-like amastigotes, Blastocrithidia moves to the Malpighian tubules, where the former species is absent (Frolov et al., Reference Frolov, Malysheva, Ganyukova, Yurchenko and Kostygov2018). A similar situation possibly occurs in the co-infection of Niesthrea sidae by Leptomonas sp. 1 and Obscuromonas sp. TU73, and Ochrostomus pulchellus by Leptomonas sp. 1 and Obscuromonas sp. 1 detected in this study.
One possible source to compete for (apart from nutrition) is the epithelial surface to which trypanosomatids tend to adhere. In such a case, species living freely in the lumen should have a higher capacity for co-existence, as this competition factor is excluded. Such species can be confined to the midgut, where attachment is impossible (except for Heteroptera) due to the presence of peritrophic matrix (Kostygov et al., Reference Kostygov, Frolov, Malysheva, Ganyukova, Chistyakova, Tashyreva, Tesařová, Spodareva, Režnarová, Macedo, Butenko, D'avila-Levy, Lukeš and Yurchenko2020). Indeed, while in the hindgut of a muscid fly (MCu02) Herpetomonas species massively predominated, there were 3 comparably large clusters of Vickermania sp. 8, Vickermania sp. 13, and ‘Muscomonas’ in the midgut. Similarly, and not exclusively, in the midgut of another muscid fly 2 non-attaching species represented by Vickermania sp. 8 and ‘Muscomonas’ coexisted in approximately the same intensity of infection.
Combining several methods, 48 trypanosomatid species belonging to 11 genera were detected in true bugs and flies, confirming previous findings that these parasites are highly diverse and ubiquitous, with most genera having cosmopolitan distribution. Thanks to the use of deep ONT sequencing, we were able to detect a surprisingly high diversity of insect trypanosomatids in Cuba. Even more importantly, this approach allowed us to determine a substantial proportion of mixed infections, with up to 8 species of these flagellates infecting a single fly host. When extrapolated to the fraction of dipteran and hemipteran diversity infected by these flagellates in the tropics, the estimate of trypanosomatid diversity may be justifiably increased by up to an order of magnitude, revealing one more facet of this unique group of parasites.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182024000453.
Data availability statement
All data is available in the manuscript or supporting material.
Acknowledgements
We thank Michal Tkoč (National Museum, Prague) for help with insect determination and Nela Gloriková for providing some illustrations.
Author contributions
JV and JL III conceived and designed the study; JV, JL III and JL IV collected and dissected insects; PK determined insects; ES, SZ, PP and OB conducted data gathering; JV, PP and OB performed data analyses; JV, SZ and JL III wrote and edited the article.
Financial support
This work was supported by grants from the Czech Grant Agency No. 21-09283S and 22-14356S (to J.L.), the ERD funds of the Czech Ministry of Education 16_019/0000759 (to J.V. and J.L.) and the Ministry of Culture of the Czech Republic DKRVO 2024–2028/5.I.a (to P.K.).
Competing interests
The authors declare there are no conflicts of interest.
Ethical standards
Not applicable.










 Open access
Open access