



Introduction
Fleas belong to Arthropoda, Insecta and Siphonaptera. They were important vector insects parasitized on warm-blooded animals. They mainly transmitted plague and murine typhus. Fleas undergo complete metamorphosis including egg, larva, pupa and adult stage. Larva fleas lived on organic matter in the nest and were in non-parasitic state, while adult fleas mainly parasitized on medium and small mammals (Krasnov et al., Reference Krasnov, Shenbrot, Mouillot, Khokhlova and Poulin2006; Eisen et al., Reference Eisen, Borchert, Mpanga and Atiku2012). As a temporary host and principal vector for plague transmission, fleas were the early warning indicator for judging plague epidemic, and also an indispensable part of plague biological community. Fleas play important role in maintaining the stability of plague natural foci (Eisen et al., Reference Eisen, Borchert, Mpanga and Atiku2012; Zhao, Reference Zhao2017). As the ‘accomplice’ of plague transmission, fleas have always attracted considerable attention. However, due to backward sequencing technology previously, genomics research has been considerably hindered. Most researches have focused only on morphological classification and pathogen, and the accurate identification and differentiation of related species and cryptic species was often challenged (Linardi and Santos, Reference Linardi and Santos2012). In the 19th century, some researchers thought that fleas were beetles according to their morphological characteristic (Crowson and Hennig, Reference Crowson and Hennig1970). In the middle and late 20th century, it was found that fleas were closely related to Mecoptera (scorpionflies) and Diptera (flies, blackfly, mosquito etc.), together constituting the group Antliophora (Kristensen and Niels, Reference Kristensen and Niels2009). In the 21st century, with the development of molecular systematics and genome sequencing technology, it was found that Siphonaptera and Mecoptera were sister groups (Chalwatzis et al., Reference Chalwatzis, Hauf, Van De Peer, Kinzelbach and Zimmermann1996; Whiting, Reference Whiting2004). In recent years, Tihelka et al. proposed to classify the order Siphonaptera into infraorder and reduce the order number of holometabolous insects to 10 orders (Tihelka et al., Reference Tihelka, Giacomelli, Huang, Pisani, Donoghue and Cai2020); and it was thought that Siphonaptera were placed into the order Mecoptera, and the order Siphonaptera and the family Nannochoristidea were sister groups. There were about 2500 species (subspecies) of Siphonaptera in the world and 650 species (subspecies) in China (Wu, Reference Wu2007). So far, only 17 flea mitogenomes in the order Siphonaptera have been sequenced. With the recent advances of high-throughput sequencing technology, more and more flea mitogenomes have been sequenced.
Amphalius spirataenius was first collected on Ochotona thibetana in 1963 (Liu et al., Reference Liu, Wu and Wu1966). In 1975, Smit gave detailed description of the morphological characteristics of male and female individuals of A. spirataenius (Smit, Reference Smit1975). This species was later collected in Qinghai, Yunnan, Sichuan and other places (Li, Reference Li1980; Ji et al., Reference Ji, Yu and Chen1981; Cai, Reference Cai1997). In the past, most studies on A. spirataenius focused on morphological structure and living habits, while the study of mitogenome was still blank. In this study, the A. spirataenius mitogenome was determined and analysed for the first time, and its morphological characteristics were described in detail. Combined with mitogenomes of the order Siphonaptera from GenBank for comparative analysis, and the appropriate outgroups were selected to construct a phylogenetic tree, which provided molecular biological genetic data for better promoting rapid and reliable identification of fleas.
Materials and methods
Specimen collection, morphological identification, DNA extraction and mitogenome sequencing
Amphalius spirataenius was collected from the body surface of Ochotona thibetana (Rodentia, Muroidea) in Deqin County, Yunnan Province, China, and species identification was done using morphological characteristics. The morphological identification was primarily based on ‘Fauna Sinica insecta Siphonaptera’ (Wu, Reference Wu2007). Specimens were preserved in EP tubes filled with 95% ethanol and stored in a refrigerator at –80°C. Hosts and specimens were deposited in Dali University. All specimens follow small mammal capture protocol and procedures and have been approved by the Animal Ethics Committee of Dali University (No. MECDU-201912-20). Specimens were subsequently sent to Shanghai Winnerbio Technology Co., Ltd. (Shanghai, China) for DNA extraction. The integrity of the obtained total DNA was detected by gel electrophoresis. Samples with intact DNA were submitted to high-throughput sequencing on the Illumina Novoseq 6000 platform; the quality of the original data was sheared; finally, clean data were obtained.
Mitogenome assembly, annotation and analysis
MitoZ 2.3 (https://doi.org/10.1101/489955) was used to de novo assemble mitogenome. The principle was that the average sequencing depth of mitogenome reads was much higher than that of nuclear genome; different Kmer parameters were set to achieve the best assembly effect. To ensure the assembly accuracy, the sequencing raw data were mapped to the assembled genome using bwa v0.7.17 (https://bio-bwa.sourceforge.net/) and samtools v0.1.20 (https://github.com/samtools/samtools/releases?page=2). The sequencing depth of the assembly result was evaluated and generally sequencing depth of 100× or higher was deemed to indicate a high level of accuracy in the assembly results. Sequence assembly was performed using Geneious Prime 11.0 (Kearse et al., Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012) software. The tRNA genes were predicted using tRNAscan SE (Chan et al., Reference Chan, Lin, Mak and Lowe2021) and ARWEN (Laslett and Canbäck, Reference Laslett and Canbäck2008), and protein-coding genes (PCGs) and rRNA genes were identified using Geneious Prime 11.0 software, BLAST (Altschul et al., Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997) and MITOS (Bernt et al., Reference Bernt, Donath, Jühling, Externbrink, Florentz, Fritzsch, Pütz, Middendorf and Stadler2013). The annotated mitogenome sequence of A. spirataenius was deposited in the GenBank (accession number: OR855715) database. PhyloSuite (Zhang et al., Reference Zhang, Gao, Jakovlić, Zou, Zhang, Li and Wang2020) was used to calculate the relative synonymous codon usage (RSCU) and nucleotide composition. Strand asymmetry was calculated: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C). CodonW was used to calculate the RSCU. Ka (the number of non-synonymous substitutions per non-synonymous site: Ka = dN = SA/LA), Ks (the number of synonymous substitutions per synonymous site: Ks = dS = SS/LS) and ω (the ratio Ka/Ks) were estimated with the software KAKS_Calculator 2.0. (Wang et al., Reference Wang, Zhang, Zhang, Zhu and Yu2010). Conserved sites and variable sites of 13 PCGs were calculated in DnaSP 6.0 and MEGA 11.0. Nucleotide diversity was calculated in DnaSP 6.0.
Phylogenetic analysis
A total of 40 species were selected to construct a phylogenetic tree, including18 flea species, 21 insect species and Philaenus spumarius (Stewart and Beckenbach, Reference Stewart and Beckenbach2005) (NC005944) as the outgroup. Phylogenetic analysis of 13 PCGs and 2 rRNA (rrnS and rnnL) genes in 40 species was performed using maximum likelihood (ML) (Stamatakis, Reference Stamatakis2006) and Bayesian inference (BI) (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003). MAFFT (Katoh et al., Reference Katoh, Misawa, Kuma and Miyata2002) was used for sequence alignment and MACSE (Ranwez et al., Reference Ranwez, Harispe, Delsuc and Douzery2011) was used for data optimization. We estimated an ML tree using IQ-Tree and the optimal partition scheme and the best model for each partition were selected under BIC. Clade support was assessed using non-parametric bootstrap with 1000 replicates. Four independent Markov chains were run for 10 million generations in BI tree. The trees were sampled every 1000 generations with the first 25% discarded as burn-in. The phylogenetic trees were visualized and edited using FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
Morphological characteristics
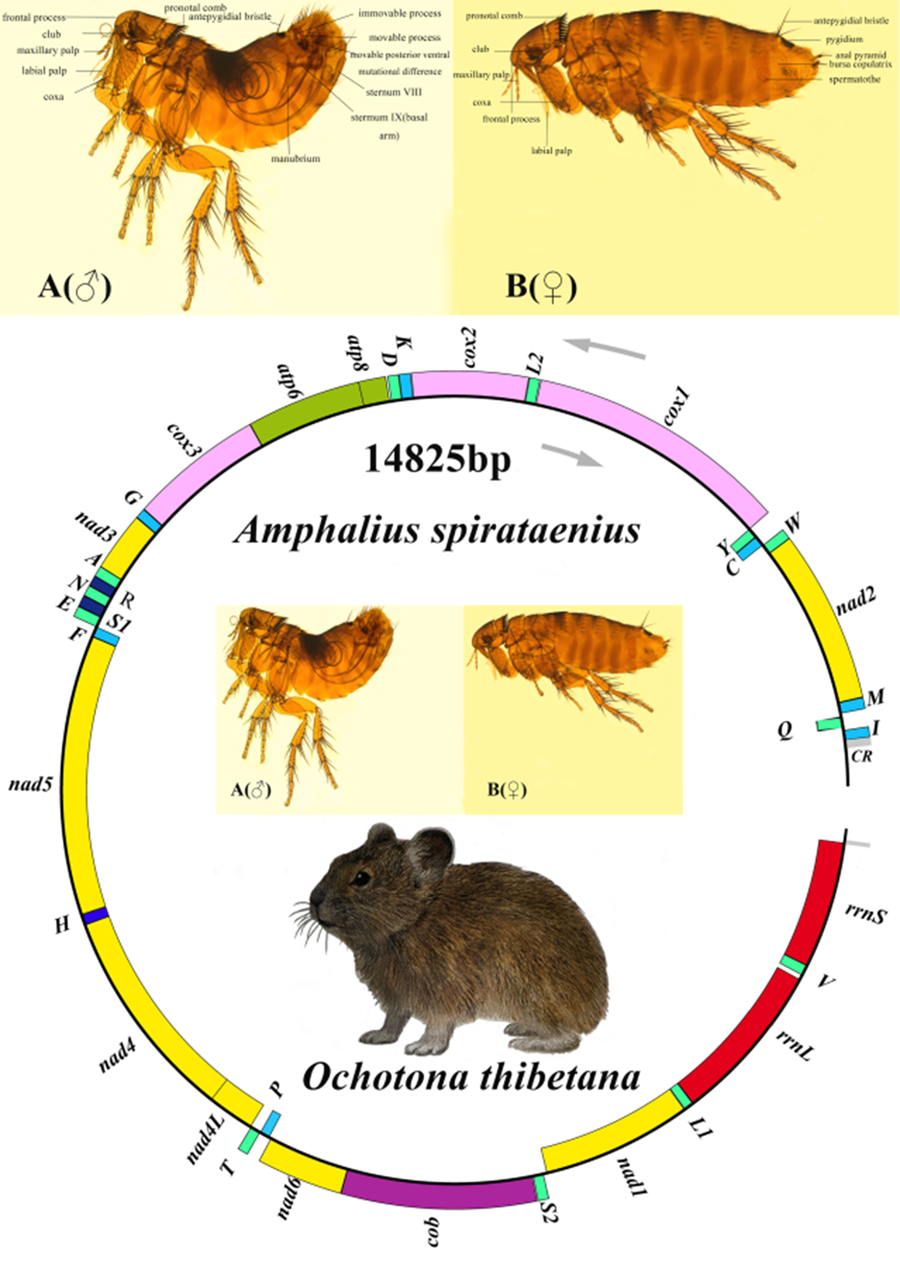
Eyes are larger; club segments with 9 tubercles, with frontal process and sharp frontal process, shaped like a tooth, slightly plunged into the forehead, situated slightly below the frontal margin; the lower labial palp must exceed the end of the coxa of the forefoot. Thorax: pronotal comb ♂ 26, ♀ 26. Abdomen: 2 bristles on each of tergum of sternum 1–7; antepygidial bristles ♂ 1, ♀ 3; ♂ apical appendage of median lamina or aedeagal apodeme curled into a spiral ribbon (the reason for the origin of the name), about 3 laps. The 9th tendon of sternum and the tendon of phallosome also curled 5 laps. The end of the process of clasper was enlarged, and the anterior and posterior horns were sharp. Movable process narrow and long, with 2 spiny bristles at the end, movable process with inverted bell-shaped base of posterior ventral process, its middle segment shorter than the end segment. ♀ Tergum ⅷ with upper lateral bristles growing radially towards the anterior superior, posterior superior and posterior inferior; has an anal pyramid with 6 bristles (Fig. S1).
Mitogenome organization
The A. spirataenius mitogenome (GenBank accession number: OR855715) was 14 825 bp, except for non-coding region (incomplete), with 37 genes of typical metazoan animals, including 13 PCGs (cox1-3, nad1-6, nad4L, atp6, atp8, cob), 22 tRNA genes and 2 rRNA genes (Table 1). Arrangement pattern of the A. spirataenius mitogenome retained that of hypothetical insect ancestors (Fig. 1). Base composition of the A. spirataenius mitogenome was A: 38.0%, T: 40.7%, G: 8.5%, C: 12.8%, with AT content of 78.7%. AT-skew was −0.034, GC-skew was −0.199. AT-skews of 13 PCGs were all negative, and GC-skews were all negative except for nad1, nad4, nad4L and nad5 (Fig. 2). The length of rrnL was 1299 bp, and the length of rrnS was 779 bp. A total of 12 intergenic regions was 68 bp. The largest intergenic region was 19 bp, followed by 16 bp and the smallest was 1 bp. There were 10 overlaps (28 bp), the largest overlapping region was 7 bp, and the smallest was 1 bp. In the A. spirataenius mitogenome, 14 genes were encoded on the N strand (trnQ, trnC, trnY, trnF, nad5, trnH, nad4, nad4L, trnP, nad1, trnL1, rrnL, trnV, rrnS), and the remaining 23 genes were encoded on the J strand.
Table 1. Distribution of the Amphalius spirataenius mitogenome


Figure 1. Organization of the Amphalius spirataenius mitogenome. tRNA genes were shown with the single-letter abbreviations of their corresponding amino acids. Note: The morphological figure of Ochotona thibetana from the volume 7 of The Mammals of The World (Wilson et al., Reference Wilson, Lacher and Mittermeier2017).

Figure 2. Skewness of 13 protein-coding genes of Amphalius spirataenius.
Protein-coding genes and codon usage bias
The total length of 13 PCGs was 11 133 bp. The total length of PCGs on the N strand was 4274 bp, and the total length of PCGs on the J strand was 6859 bp. The longest PCG was nad5 (1714 bp), and the shortest was atp8 (177 bp). All PCGs began with the typical ATN as start codon, of which nad2, nad3 and nad6 use ATT as start codon, cox1 and atp8 use ATC as start codon, nad1 uses ATA as start codon, and the remaining PCGs use ATG as start codon. Incomplete stop codon ‘T’ for nad2, nad4 and nad5, while nad3 use TAG as stop codon. The remaining 8 PCGs use TAA as complete stop codons (Table 1). The RSCU was calculated for the A. spirataenius mitogenome (Table 2, Fig. 3). The most frequently used codons were UUA (Leu) and UCU (Ser), while UCG (Ser) and ACG (Thr) were the least frequently used codons. Twenty-six codons of UUU, UUA, AUU, AUA, UAU, AAU, etc., were preference codons (RSCU > 1).
Table 2. Codon usage of protein-coding genes in the Amphalius spirataenius mitogenome


Figure 3. Relative synonymous codon usage (RSCU) of Amphalius spirataenius. The Y-axis represents the RSCU value, and the X-axis represents the codons corresponding to each amino acid.
RSCU was mainly measured to know the relative probability of specific codon in the synonymous codon encoding the corresponding amino acid, which can intuitively reflect codon usage preference, but the reason for its preference was not clear. Parity rule 2 (PR2) (Sueoka, Reference Sueoka1995), effective codon count (ENC-plot) (Wright, Reference Wright1990) and neutrality curve (Patil et al., Reference Patil, Indrabalan, Suresh and Shome2021) analyses of A. spirataenius were performed to evaluate factors that influenced evolutionary processes. PR2 indicated by qualitative analyses that mutation, selection and other factors together influence codon usage, and it was concluded that the third base of codons has T/C preference (Fig. 4A). In ENC-plot, except for the Nc values of nad4L (37.19) and nad6 (36.87) > 35, the remaining PCGs were lower than 35 (Nc < 35) and most of them fall below standard curve (red) (Fig. 4B). Neutral curve indicated that regression coefficient was –0.736, and all values were above diagonal curve (red) (Fig. 4C).

Figure 4. Analysis of 13 protein-coding genes of Amphalius spirataenius. (A) PR2; (B) ENC-plot; (C) neutral curve.
The evolutionary rates of 13 PCGs were calculated and analysed using KAKS_Calculator2.016 software with Drosophila Yakuba (Clary and Wolstenholme, Reference Clary and Wolstenholme1984) as the outgroup. Among 13 PCGs, nad6 (Ka/Ks = 0.272) had the fastest evolutionary rate, followed by atp8 (Ka/Ks = 0.167). Evolution rate of cox1 (Ka/Ks = 0.024) was the slowest. The ratio rate (Ka/Ks) of 13 PCGs was lower than 1 (Fig. S2). The proportion of conserved sites among 13 PCGs was the highest for cox1 (0.809), lowest for atp8 (0.598) and second for nad6 (0.636). The percentage of variant sites for nad2 (0.329) was the highest, second lowest for nad6 (0.318) and lowest for cox1 (0.190). The nucleotide diversity of atp8 gene was 0.345, which was the most diverse nucleotide among all PCGs genes, followed by nad2 (Pi = 0.337) and nad6 (Pi = 0.333) (Table S2, Fig. S2).
tRNA and rRNA gene analysis
Among 22 tRNA genes, the longest was 70 bp (trnK and trnT), the shortest was 62 bp (trnL1 (tga)), and the average length was 65.2 ± 2.7 bp. The anticodons of trnK (CUU) and trnS1 (UCU) in A. spirataenius were different from those of some other arthropod mitochondrial trnK (UUU) and trnS1 (GCU) (Sun et al., Reference Sun, Chen and Dong2022; Yang et al., Reference Yang, Yang, Ren and Dong2023; Yuan et al., Reference Yuan, He and Dong2023). During tRNA gene folding, in addition to the typical Watson–Crick (A–U, G–C) pairing, there were 19 mismatch base pairs, of which there were 15 G–U mismatches, 2 U–U mismatches, 1 C–A mismatch and 1 C–U mismatch. The remaining 21 tRNA genes formed the typical cloverleaf structures, except for trnS1 which lacked the D-arm (Fig. S3). The lengths of 2 rRNA genes were 1299 bp (rrnL) and 779 bp (rrnS) respectively, where AT/GC content of rrnL gene was 81.9%/12.3%, while that of rrnS gene was 80.4%/12.7%, both of which were encoded on the N strand (Table 1).
Phylogenetic analysis
Concatenated nucleotide sequences of 13 PCGs and 2 rRNA genes (13 PCGs + 2 rRNAs) from the mitogenomes of 40 insect species (18 flea species of Siphonaptera) were analysed using BI and ML method to construct a phylogenetic tree with P. spumarius (NC005944) as outgroup. The 2 phylogenetic tree topologies are slightly different, but both have high node support (posterior probabilities >95%, bootstrap values >70%) (Figs 5 and 6). The phylogenetic tree the hypothesis supported that the order Siphonaptera is monophyly and formed the major clade with 3 branches, with the Bayesian posterior probability (Bpp = 1) and the Ultrafast bootstrap approximation value (UFBoot = 100%) in the BI and ML analyses, respectively.

Figure 5. Phylogenetic tree of 40 insect species was constructed using Bayesian methods with Philaenus spumarius as the outgroup and node values as posterior probability values (PP). Amphalius spirataenius was labelled in red.

Figure 6. Phylogenetic tree of 40 insect species was constructed by maximum likelihood method with Philaenus spumarius as an outgroup and node values as bootstrap values (BS). Amphalius spirataenius was labelled in red.
Discussion
The A. spirataenius mitogenome was reported for the first time in this study. The 14 825 bp long mitogenome has the typical metazoan of 37 genes with insect ancestral genome arrangement patterns (Clary and Wolstenholme, Reference Clary and Wolstenholme1984). To date, the mitogenomes of 18 flea species have been sequenced in the world and their AT/GC contents are shown in Table S1. The AT content of fleas is as high as 76.7–83.2%, which is much higher than that of parasitic lice (Polyplax asiatica AT = 58%, Polyplax spinulosa AT = 61%) (Zhang and Dong, Reference Zhang and Dong2020) and the 2 orders Mecoptera and Diptera (Song et al., Reference Song, Li, Jiang, Zhou, Liu, Sun, Vogler and Cai2016a, Reference Song, Chen, Wei and Chen2016b). High AT content may be the main reason for incomplete sequencing or sequencing failure of control regions. AT-skew indicated that the remaining 17 flea species had negative AT-skew except for Leptopsylla segnis (AT-skew = 0.024) and Neopsylla specialis (AT-skew = 0), while GC-skew was positive for the remaining 17 flea species except for Leptopsylla segnis (GC-skew = 0.248), which might be resulted from directional evolutionary pressure and asymmetric replication. It might be also related to living environment and parasitic life history because only adult fleas parasitize on host. Negative GC-skew in the J-strand and positive GC-skew in the N-strand of A. spirataenius were consistent with the mitogenome of most metazoan (Dermauw et al., Reference Dermauw, Van Leeuwen, Vanholme and Tirry2009).
There are 12 intergenic spacers in the A. spirataenius mitogenome, with the largest intergenic spacer (19 bp) located between trnS2 and nad1 (Table 1). Intergenic spacer might be transcription termination signal site of transcription process (Cameron and Whiting, Reference Cameron and Whiting2008; Yang et al., Reference Yang, Yang, Ren and Dong2023). There were 10 gene overlaps, with the largest overlap region of 7 bp (atp6 and atp8; nad4 and nad4L) and the overlap region between atp6 and atp8 is common in arthropod mitogenomes (Ge et al., Reference Ge, Liu, Kang, Liu and Yang2022). Some researchers believed that gene intergenic and overlap regions of mitogenome might be beneficial for mitochondrial structural stability (Song et al., Reference Song, Li, Jiang, Zhou, Liu, Sun, Vogler and Cai2016a, Reference Song, Chen, Wei and Chen2016b).
The 13 PCGs of A. spirataenius had typical ATN as the start codon. In fact, the start codons of 13 PCGs in some metazoans are not entirely typical ATNs. For example, atp8 of Eulaelaps huzhuensis used GTG as the start codon (Yang et al., Reference Yang, Yang, Ren and Dong2023). Most PCGs of Mecoptera and Diptera used TCG as the start codon (Clary and Wolstenholme, Reference Clary and Wolstenholme1984; Beckenbach, Reference Beckenbach2011), and the cox1 of some insects used GCA as the start codon (Cameron, Reference Cameron2015). It was often the case that cox1 of insects had atypical start codons, most likely due to the 1 bp deletion that caused the TCG frameshift (Cameron, Reference Cameron2015). Nad2, nad4 and nad5 of A. spirataenius had incomplete stop codon ‘T’, while nad3 used TAG as stop codon. The remaining 8 PCGs used the typical TAN as stop codon. PCGs of many arthropods use incomplete stop codons (Liu et al., Reference Liu, Yu, Yu, Bi, Yang, Xue, Zhang, Zhang, Yi, Ma, Zhou, Lan, Gu, Wu, Li and Qi2022). These incomplete stop codons might be transcribed by polyadenylation to obtain the complete stop codon TAA (Huang et al., Reference Huang, Chen, Wei and Shi2022).
We calculated the RSCU for 13 PCGs of A. spirataenius for codon usage preference assessment (Fig. 3). Twenty-six codons of UUU, UUA, AUU, AUA, UAU, AAU, etc., were preference codons (RSCU > 1), RSCU > 1 indicated relatively high codon usage frequency (Liu et al., Reference Liu, Zhu, Ma, Liu, Wang, Jia, Chen, Sun, Yang, Wu, Chen and Cheng2016; Yang et al., Reference Yang, Yang, Ren and Dong2023). Most codons ending with A/U bases were frequently used, while codons ending with G/C were rarely used, or even in some cases not used in the A. spirataenius mitogenome, which was consistent with the RSCU of other metazoan (Hao et al., Reference Hao, Zou, Ding, Xu, Yan, Li, Fu, Li and Chen2017). There were some exceptions, such as Diptera's preference for codons ending in G/C (Vicario et al., Reference Vicario, Moriyama and Powell2007; Behura and Severson, Reference Behura and Severson2011). Generally, codon usage patterns were more similar between closely related species. In order to evaluate factors that influenced evolutionary processes, PR2 (Fig. 4A), ENC-plot (Fig. 4B) and neutral curve analyses (Fig. 4C) of A. spirataenius were performed. PR2, ENC-plot and neutrality curve analyses indicated that the influence of codon usage preference of A. spirataenius might be mainly resulted from selection pressure. Additionally, base composition (Arhondakis et al., Reference Arhondakis, Auletta, Torelli and D'Onofrio2004), overall expression level of gene (Hiraoka et al., Reference Hiraoka, Kawamata, Haraguchi and Chikashige2009), nature of amino acids (aromatic and hydrophobic) (Knight et al., Reference Knight, Freeland and Landweber2001) and codon context (Jia and Higgs, Reference Jia and Higgs2008) might also be influenced by codon usage preference.
The evolutionary rates of 13 PCGs were calculated and analysed using KAKS_Calculator2.016 software. Ka/Ks < 1 of 13 PCGs of A. spirataenius indicated that PCGs were subject to negative or purifying selection, and slow evolution. Combined with Ka/Ks, conserved sites, variable sites and nucleotide diversity analysis, it was found that nad6 had the fastest evolutionary rate, followed by atp8 and nad2 in the A. spirataenius mitogenome. Cox1 had the slowest evolution rate and is suitable for phylogenetic analysis or species classification. In 2023, Jakovlić et al. proposed that the evolutionary rates of bilaterally symmetrical animals are in descending order: internal parasites > weakly motile ectoparasites > weakly motile and free-living animals > parasitoid lineages > strongly motile ectoparasites > micropredators and strongly motile free-living animal (Jakovlić et al., Reference Jakovlić, Zou, Ye, Zhang, Liu, Xiang, Wang and Zhang2023). Fleas are strongly motile ectoparasites that may have been subjected to stronger selective pressures and so have evolved at lower rate, whereas some ectoparasites may have undergone accelerated evolution due to slack selective pressures, where some mutations had no effect on its life activities. The reason for the slow evolutionary rate of fleas may also be related to their life history, parasitism and metabolic rate in the body. Species evolution was a complex and variable process, and apart from the above possible reasons, there are some other factors such as generation time, replication and repair machinery, directional selection driven by host parasite arms race, etc. (Dawkins and Krebs, Reference Dawkins and Krebs1979; Haraguchi and Sasaki, Reference Haraguchi and Sasaki1996; Jakovlić et al., Reference Jakovlić, Zou, Ye, Zhang, Liu, Xiang, Wang and Zhang2023).
The average length of 22 tRNA genes of A. spirataenius was 65.2 ± 2.7 bp, which was longer than that of Parasitiformes (62.0 ± 1.3 bp) (Yuan et al., Reference Yuan, Wei, Wang, Dou and Wang2010). The anticodon of trnK (CUU) was different from that of some other arthropod trnK (UUU) (Sun et al., Reference Sun, Chen and Dong2022; Yuan et al., Reference Yuan, He and Dong2023); trnS1 used UCU as an anticodon, whereas trnS1 of most arthropods used GCU as an anticodon (Yang et al., Reference Yang, Yang, Ren and Dong2023). The secondary structures of 22 tRNA genes of A. spirataenius are shown in Fig. S3; of which 21 genes had typical cloverleaf structure except for trnS1 which lacked the D-arm. Generally, trnS1 which lacked the D-arm was prevalent in metazoan (Wang et al., Reference Wang, Liu, Winterton and Yang2012). The tRNA secondary structure might be related to species evolution (Watanabe et al., Reference Watanabe, Kawai, Yokogawa, Hayashi, Kumazawa, Ueda, Nishikawa, Hirao, Miura and Watanabe1994). The tRNA secondary structure of A. spirataenius had 19 mismatch base pairs, of which there were 15 G–U mismatches, 2 U–U mismatches, 1 C–A mismatch and 1 C–U mismatch. G–U mismatches were favourable to the maintenance of the tRNA secondary structure (He and Dong, Reference He and Dong2023) and G–U mismatches mostly occurred at turn point of the tRNA secondary structure.
The phylogenetic tree demonstrated that A. spirataenius, Ceratophyllus anisus, Ceratophyllus wui, Paradoxopsvllus custodis and Jellisonia amadoi were sister groups (PP = 1, BS = 100) and species of the same family or genus with well-defined taxonomic status were clustered together. It was consistent with the traditional morphological classifications. Ceratophyllidae and Leptopsyllidae were clustered into 1 major clade, while Pulicidae, Ctenophthalmidea, Hystrichopsylloidea and Vermipsyllidae were clustered into the other major clade. Ceratophyllidae formed sister group with the other 5 families. The families Ceratophyllida, Leptopsyllidae, Ctenophthalmidea and Hystrichopsylloidea were paraphyletic. Interestingly, Dorcadia ioffi were clustered with Hystrichopsylla weida qinlingensis with high support (PP = 1.0, BS = 95), which was somewhat inconsistent with the view of Wu (Reference Wu2007). The superfamily Vermipsylloidea and Hystrichopsylloidea were separated from Ceratophylloidea forming 2 independent families respectively. Interestingly, 3 superfamilies had obvious differences in morphology, but belong to the same clade in the phylogenetic tree. Phylogenetic relationship will be further explored by enlarged sample size in the future.
The phylogenetic tree strongly supported that the orders Siphonaptera and Mecoptera were monophyly. It was accordant to previous studies (Misof et al., Reference Misof, Liu, Meusemann, Peters, Donath, Mayer, Frandsen, Ware, Flouri, Beutel, Niehuis, Petersen, Izquierdo-Carrasco, Wappler, Rust, Aberer, Aspöck, Aspöck, Bartel, Blanke, Berger, Böhm, Buckley, Calcott, Chen, Friedrich, Fukui, Fujita, Greve, Grobe, Gu, Huang, Jermiin, Kawahara, Krogmann, Kubiak, Lanfear, Letsch, Li, Li, Li, Lu, Machida, Mashimo, Kapli, McKenna, Meng, Nakagaki, Navarrete-Heredia, Ott, Ou, Pass, Podsiadlowski, Pohl, von Reumont, Schütte, Sekiya, Shimizu, Slipinski, Stamatakis, Song, Su, Szucsich, Tan, Tan, Tang, Tang, Timelthaler, Tomizuka, Trautwein, Tong, Uchifune, Walzl, Wiegmann, Wilbrandt, Wipfler, Wong, Wu, Wu, Xie, Yang, Yang, Yeates, Yoshizawa, Zhang, Zhang, Zhang, Zhang, Zhao, Zhou, Zhou, Ziesmann, Zou, Li, Xu, Zhang, Yang, Wang, Wang, Kjer and Zhou2014; Meusemann et al., Reference Meusemann, Trautwein, Friedrich, Georg, Wiegmann, Donath, Podsiadlowski, Petersen, Niehuis, Mayer, Bayless, Shin, Liu, Hlinka, Minh, Kozlov, Morel, Peters, Bartel, Grove, Zhou, Misof and Yeates2020). The order Hymenoptera was the earliest clade. In recent years, some researchers suggested that the order Siphonaptera formed a sister group with the family Nannochoristidea, and the order Siphonaptera was treated as a member (the infraorder Siphonaptera) of the order Mecoptera (Tihelka et al., Reference Tihelka, Giacomelli, Huang, Pisani, Donoghue and Cai2020). However, the family Nannochoristidea and the order Siphonaptera were a separate clade, respectively, in the phylogenetic tree. The order Siphonaptera was closely related to the orders Neuroptera, Diptera and Mecoptera and formed a sister group (PP = 0.95, BS = 60) (Figs 5 and 6). These results have provided new insights into the phylogenetic position of the order Siphonaptera within holometabolous insects. However, in current study, all lineages of fleas were not included in the analyses. Therefore, further study involving more mitogenomes of all flea families in the order Siphonaptera was needed to reassess phylogenetic relationship of the order Siphonaptera within holometabolous insects and obtain more reliable results.
Conclusion
Organization and evolution of the A. spirataenius mitogenome were reported for the first time in this study. It provided unique insights into the phylogenetic and taxonomic status of the order Siphonaptera, which were monophyletic and belong to order level rather than infraorder level. The A. spirataenius mitogenome provides new molecular data for phylogeny and taxonomic level of the order Siphonaptera. To obtain a more reliable phylogenetic tree, we still need to collect more representative species of the order Siphonaptera and to sequence the mitogenomes of more species, and to study evolutionary mechanisms of the order Siphonaptera in depth.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182024000635.
Data availability statement
All data generated or used during the study appear in our manuscript.
Acknowledgements
We thank Ting Chen and Rong Fan for his help in specimen collection.
Author contributions
Ju Pu and Wenge Dong designed and performed the research; Ju Pu and Wenge Dong contributed reagents and materials. Wenge Dong, Ju Pu and Xiaoxia Lin analysed the data; Ju Pu and Wenge Dong wrote the manuscript. All the authors have read and approved the final manuscript.
Financial support
We acknowledge funding support from the National Natural Science Foundation of China (No. 32260152 to Wenge Dong).
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical standards
All methods and procedures used in the rodent capture process were in accordance with the guidelines and regulations approved by the Animal Ethics Committees at Dali University. Approval ID is MECDU-201912-20.











 Open access
Open access