


Introduction
CVD, particularly CHD, remains a dominant cause of morbidity/mortality globally. Nonetheless, CHD is largely preventable, involving modifiable factors such as poor diet and physical inactivity, associated dyslipidaemia and obesity, smoking, psychosocial stressors and socio-economic adversity. Recently there has been considerable focus on the role of the body’s microbiota in non-communicable diseases. The gut microbiota may be a key nexus linking major modifiable risk factors (diet, obesity, inactivity, stress) to diverse chronic diseases(Reference Blum1), with trimethylamine (TMA) N-oxide (TMAO) potentially participating in these linkages(Reference Wang, Klipfell and Bennett2) (Fig. 1). Indeed, evidence that dietary lipids are not a major CVD risk factor(Reference Siri-Tarino, Sun and Hu3) encourages investigation of such alternative diet-linked drivers of disease.
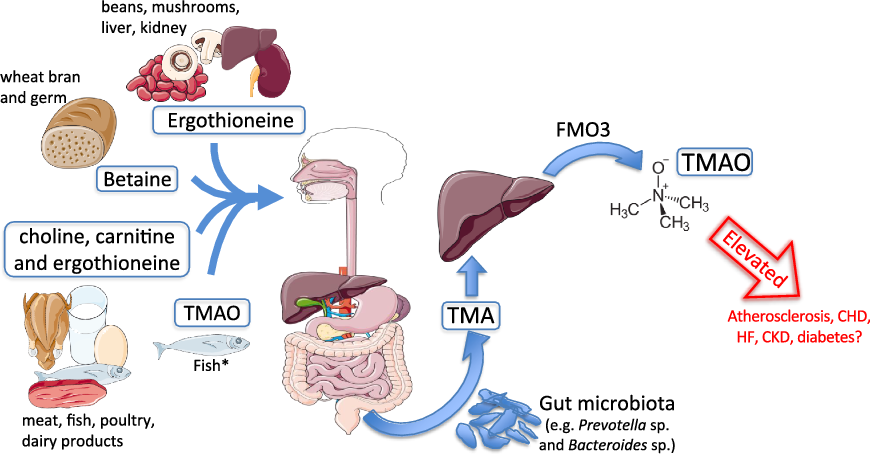
Fig. 1. Trimethylamine-N-oxide (TMAO) formation. Process of TMAO intake or generation from dietary sources. Substantial elevations in circulating TMAO are hypothesised to promote cardiometabolic and renal diseases. * Fish represent a significant and direct dietary source of TMAO. CKD, chronic kidney disease; FMO3, flavin-containing mono-oxygenase enzyme 3; HF, heart failure; TMA, trimethylamine.
Bolstered by evidence of detrimental effects of high TMAO concentrations in cell(Reference Mohammadi, Vahabzadeh and Jamalzadeh4-Reference Chen, Zhu and Ran7) and animal models(Reference Li, Chen and Gua8-Reference Zhang, Meng and Yu13), recently reported associations between TMAO and CVD risk and outcomes(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14-Reference Mente, Chalcraft and Ak17) have focused attention on TMAO as a potential determinant of disease. This may provide both a circulating biomarker of CVD risk, and a potential therapeutic target (Fig. 2). Nonetheless, whether the effects and disease associations of TMAO reflect a primary causal or secondary reinforcing role in CVD is unclear, and critical unknowns remain unresolved: how are TMAO levels elevated, and is this a consequence or cause of disease; what do association studies indicate in terms of the role of TMAO in CVD; are associations consistent with current knowledge regarding TMAO pathogenicity? More broadly, is a primary role for TMAO reconcilable with known dietary risks for and determinants of CVD?

Fig. 2. Trimethylamine-N-oxide (TMAO) responses in health and diseased omnivores. Variations in circulating TMAO as a result of dietary substrates in omnivores may be insufficient to promote disease. However, a combination of major risk factors (ageing, obesity, insulin resistance, shift in gut microbiota) and/or disease (diabetes, chronic kidney disease) – as in increasingly common multimorbidity – may cumulatively and chronically elevate circulating TMAO to >10–20 µm. These concentrations may be sufficient to promote vascular and cardiac dysfunction/disease. Speculative positive feedbacks are also presented, based on putative roles of TMAO in inflammation, cardiac and vascular dysfunction, and flavin-containing mono-oxygenase 3 (FMO3) activity: evolving CVD can induce renal dysfunction to elevate TMAO; inflammation promotes TMAO generation and FMO3 expression; and TMAO itself together with renal dysfunction may up-regulate FMO3 activity. These putative feedbacks await experimental confirmation. RCT, reverse cholesterol transport; SR, scavenger receptor; TMA, trimethylamine.
Pathogenic effects of trimethylamine N-oxide
The pathogenic effects and potency of TMAO are fundamental to its potential role in CVD. Evidence to date supports detrimental effects of high TMAO levels on atherosclerotic processes, and vascular and myocardial function, a suite of actions that could well promote CVD. However, there are inconsistencies between the concentration dependence of TMAO effects in experimental systems and pathogenic sensitivities implied in some TMAO–CVD associations: the latter suggest markedly increased CVD risk and complications with <5 µm TMAO(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Mente, Chalcraft and Ak17-Reference Sun, Jiao and Ma19) , despite little direct evidence for detrimental effects at these concentrations. Indeed, pathological concentrations in experimental systems may be rarely encountered in vivo, except in advanced CVD or renal disease.
Pro-atherosclerotic actions
There has been a focus on roles of TMAO in atherosclerosis and vascular dysfunction. At substantially elevated concentrations, TMAO is pro-inflammatory, impairs vascular function and structure, and may up-regulate scavenger receptors (SR) while inhibiting reverse cholesterol transport (RCT)(Reference Seldin, Meng and Qi6,Reference Chen, Zhu and Ran7,Reference Sun, Jiao and Ma19,Reference Koeth, Wang and Levison20) (Fig. 3). High TMAO concentrations can also induce platelet hyper-reactivity, enhancing thrombotic potential via amplified intracellular Ca2+ release(Reference Zhu, Gregory and Org21). A 10-fold elevation in serum TMAO with choline supplementation in mice (1 % w/w, about 12× normal) also augments platelet reactivity to ADP, though failing to influence other indices including surface phosphatidylserine content in ADP-stimulated platelets, levels of Von Willebrand factor, α granule release, or baseline pro-thrombotic microvesicle release(Reference Roberts, Gu and Buffa22). These mixed actions could contribute to vascular disease when TMAO exceeds 10–20 µm, for example in heart failure (HF) and chronic kidney disease (CKD); however, involvement in the initial pathogenesis of atherosclerosis and CHD is questionable(Reference Wang, Roberts and Buffa23).
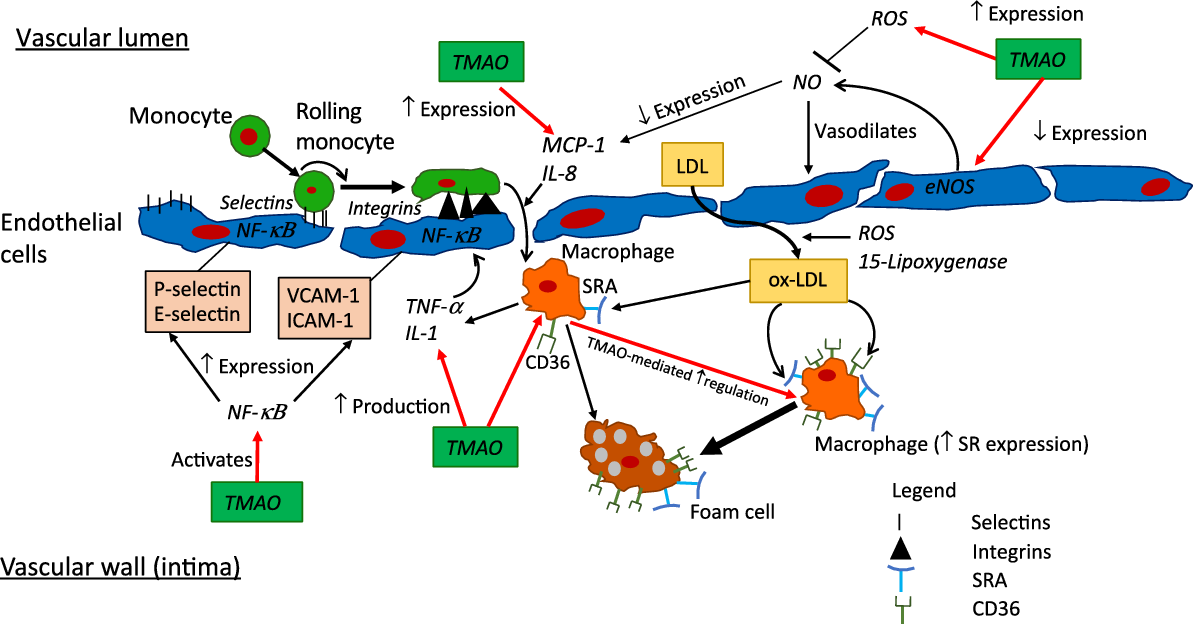
Fig. 3. Putative pro-inflammatory and -atherosclerotic actions of trimethylamine-N-oxide (TMAO). Pronounced elevations in TMAO to >10–20 µm, for example in advanced heart failure or chronic kidney disease, may be sufficient to modify multiple determinants of inflammation and atherosclerosis, as detailed here. Whether levels of circulating TMAO in obesity, diabetes or CHD are sufficient to significantly influence these processes is presently unclear. CD36, cluster of differentiation 36; eNOS, endothelial nitric oxide synthase; ICAM-1, intracellular adhesion molecule-1; MCP-1; monocyte chemoattractant protein 1; NF-κB; nuclear factor κ-light-chain-enhancer of activated B cells; ox-LDL, oxidised-LDL; ROS, reactive oxygen species; SR, scavenger receptor; SRA, scavenger receptor A, VCAM-1, vascular adhesion protein-1.
Studies in genetic models predisposed to disease indicate that major elevations in TMAO with high-level carnitine supplementation (1–1·3 % in drinking water) promote atherogenesis. Carnitine supplementation exaggerates atherosclerosis in ApoE–/– mice, an effect countered by broad-spectrum antibiotics(Reference Koeth, Wang and Levison20) and the choline analogue inhibitor of TMA lyase, 3,3-dimethyl-1-butanol (DMB)(Reference Wang, Roberts and Buffa23). Pro-atherosclerotic effects of intermittent hypoxia/hypercapnia (relevant to sleep apnoea) in ApoE–/– and LDL receptor knockout mice are also partially DMB sensitive(Reference Xue, Zhou and Poulsen24). These observations support some role for TMAO in high-risk settings where both propensity to atherosclerosis and TMAO concentrations are artificially augmented; however, their broader relevance is less clear. A word of caution is also warranted regarding the applicability of the ApoE–/– model to human atherosclerosis, given lack of the cholesteryl ester transfer protein important in human RCT. Contrasting these studies, analysis of outcomes in cholesteryl ester transfer protein-transfected ApoE–/– mice supports a protective rather than detrimental role for TMAO(Reference Collins, Drazul-Schrader and Sulpizio25). Other work reveals no relationship between TMAO, plasma cholesterol and atherogenesis in mice fed normal diets(Reference Veeravalli, Karu and Scott26), or in ApoE–/– mice fed high-fat diets(Reference Geng, Yang and Wang27). Differences in TMAO within inbred strains of mice are also estimated to explain less than 10 % of the variance in atherosclerosis(Reference Bennett, de Aguiar Vallim and Wang28).
Up-regulation of scavenger receptors
Increased expression of atherogenic SR, for example cluster of differentiation 36 (CD36) and scavenger receptor A (SR-A), is a putative mechanism for TMAO-sensitive atherogenesis(Reference Wang, Klipfell and Bennett2,Reference Koeth, Wang and Levison20,Reference Geng, Yang and Wang27,Reference Velasquez, Ramezani and Manal29) (Fig. 2 and Fig. 3). The membrane glycoprotein CD36 is essential in plaque formation, internalising oxidised LDL within the vascular intima(Reference Kunjathoor, Febbraio and Podrez30,Reference Ben, Zhu and Zhang31) , while SR-A1 is key to macrophage cholesterol accumulation/foam cell formation, and modulates immune function, cell proliferation and death(Reference Kunjathoor, Febbraio and Podrez30,Reference Ben, Zhu and Zhang31) . Importantly, effects of TMAO on macrophage SR-A1 expression are only evident at exceedingly high in vitro concentrations (≥300 µm)(Reference Wang, Klipfell and Bennett2,Reference Mohammadi, Vahabzadeh and Jamalzadeh4,Reference Mohammadi, Najar and Yaghoobi5) , unlikely to be encountered in vivo. Macrophage stress responses are similarly evident at very high TMAO concentrations(Reference Mohammadi, Najar and Yaghoobi5). Macrophage CD36 and SR-A1 expression in atherosclerotic mice are increased in response to an excessive 350 µm plasma concentration (achieved with TMAO supplementation)(Reference Wang, Klipfell and Bennett2,Reference Mohammadi, Vahabzadeh and Jamalzadeh4,Reference Mohammadi, Najar and Yaghoobi5) . Geng et al. (Reference Geng, Yang and Wang27) recently showed that TMAO supplementation (1 mm in drinking water) increases plaque CD36 in ApoE–/– mice, and that 100 µm TMAO exaggerates oxidised LDL-dependent changes in CD36 and foam cell formation (via p38 MAPK/JNK1/2 signalling) without influencing baseline CD36 expression(Reference Geng, Yang and Wang27). These data suggest a possible role for high concentrations of TMAO in exaggerating SR changes in advanced disease, without effects in otherwise healthy animals. Nonetheless, the causal involvement of SR-A1 or CD36 in TMAO-sensitive atherosclerosis has yet to be established.
Inhibition of reverse cholesterol transport
Additional to promoting sequestration of oxidised LDL, elevated TMAO may inhibit the RCT that underlies hepatic catabolism and biliary excretion of lipids(Reference Cucuianu, Coca and Hancu32) (Fig. 2). Dietary supplementation with carnitine (1·3 %), choline (1·0 %) or TMAO (0·12 %) decreases RCT in mice, with effects of carnitine and choline eliminated by antibiotics(Reference Wang, Klipfell and Bennett2,Reference Koeth, Wang and Levison20) . The basis of this effect awaits clarification; however, shifts in transporters such as ATP-binding cassette transporter A1 (ABCA1) and scavenger receptor B1 (SR-B1) could contribute. High concentrations of TMAO (75–150 µm) may reduce macrophage ABCA1 expression in vitro (Reference Mohammadi, Najar and Yaghoobi5), although others report increased macrophage ABCA1 with 100 µm TMAO, no change in SR-B1 with 50–100 µm TMAO, and no changes in hepatic SR-B1 protein or ABCA1 mRNA with TMAO supplementation in atherosclerotic mice(Reference Koeth, Wang and Levison20). Similarly excessive concentrations of TMAO (50–200 µm) moderately increase ABCA1-dependent cholesterol efflux(Reference Wang, Klipfell and Bennett2,Reference Koeth, Wang and Levison20) , while Trenteseaux et al. (Reference Trenteseaux, Gaston and Aguesse33) report opposing increases in SR-B1 mRNA in male offspring of hypercholesterolaemic mice v. decreases in female offspring. Other effects of TMAO that might influence RCT and atherosclerosis include reductions in bile acid pool size, synthetic enzymes and transport proteins(Reference Koeth, Wang and Levison20), including Cyp7a1, a major rate-limiting synthetic enzyme in cholesterol catabolism(Reference Pullinger, Eng and Salen34). Potential modulation of RCT by TMAO requires further study, and causal roles for ABCA1 or SR-B1 in TMAO-dependent changes in RCT and atherosclerosis remain to be demonstrated.
Pro-inflammatory actions
Inflammation is important in atherogenesis, vascular dysfunction and remodelling(Reference Ross35,Reference Wong, Meredith and Lin36) , and participates broadly in cardiometabolic dysfunction and disease. Studies in vitro and in animal models indicate that elevated TMAO can be pro-inflammatory(Reference Seldin, Meng and Qi6-Reference Li, Chen and Gua8,Reference Sun, Jiao and Ma19,Reference Geng, Yang and Wang27) , although again the concentrations required are up to an order of magnitude higher than observed in different disease states. Sufficiently high TMAO concentrations may influence multiple determinants of inflammation and associated vascular dysfunction, including integrin expression, reactive oxygen species (ROS) generation, NF-κB signalling and cytokine production, the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, and anti-inflammatory sirtuins (Fig. 3). Consistent with pro-inflammatory effects of age-dependent elevations in TMAO, inhibiting TMA formation limits changes in TNF-α, IL-1β and endothelial NO synthase (eNOS) in ageing rats (in which TMAO was elevated >2-fold), whereas expression was unaltered in young animals(Reference Seldin, Meng and Qi6,Reference Li, Chen and Gua8) .
Sun et al. (Reference Sun, Jiao and Ma19) report concentration- and time-dependent effects of 100–300 µm TMAO on IL-1β, IL-18, NLRP3 and oxidative stress in human endothelial cells, with eNOS expression and NO release repressed. Chen et al. (Reference Chen, Zhu and Ran7) also document increased human endothelial expression of IL-1β and NLRP3, together with ICAM-1, matrix metallopeptidase-9 (MMP-9), and caspase-1, on exposure to extremely high (300–900 µm) TMAO. These effects, together with NLRP3 inflammasome activation, appear related to decreased superoxide dismutase 2 (SOD2) and sirtuin-3, and increased mitochondrial ROS generation(Reference Chen, Zhu and Ran7). Similar sensitivities are reported in rat vascular smooth muscle cells, with IL-1β, NF-κB and NLRP3 increased in response to 100–600 µm TMAO(Reference Zhang, Li and Yang37). Expression of TNF-α, IL-6 and intercellular adhesion molecule 1 (ICAM-1) are also increased by TMAO in the ApoE–/– model and in cultured macrophages, facilitating macrophage recruitment; however, the required TMAO concentrations were unreported(Reference Geng, Yang and Wang27).
Choline supplementation (1·3 % in drinking water) to increase plasma TMAO >10-fold (about 55 µm) significantly increases vascular transcription of inflammation-related factors in the atherosclerosis-prone LDL receptor knockout mouse, including monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), TNF-α, ICAM-1, keratinocyte chemoattractant (mouse analogue of IL-8), cyclo-oxygenase-2 (COX-2), E-selectin, vascular adhesion protein-1 (VCAM-1) and the macrophage marker CD68(Reference Seldin, Meng and Qi6). Effects in wild-type mice were not assessed, while even higher TMAO concentrations (100–200 µm) were found to increase inflammatory mediators in human vascular endothelial and smooth muscle cells, with 400 µm TMAO increasing endothelial recruitment of leucocytes(Reference Seldin, Meng and Qi6). These effects were suppressed by inhibitors of NF-κB and the βγ G-protein subunit of G-protein coupled receptors (GPCR)(Reference Seldin, Meng and Qi6), though whether the latter reflects direct TMAO–GPCR interactions or modulation of protein conformation is unclear(Reference Koeth, Wang and Levison20). Interaction between TMA and GPCR has been reported(Reference Suska, Ibanez and Lundstrom38). Contrasting evidence of pro-inflammatory actions, TMAO appears to have no effect on toll-like receptor 4 signalling(Reference Sun, Jiao and Ma19). Overall, while some studies support pro-inflammatory effects of high TMAO levels, these concentrations are only relevant to advanced HF and CKD, whereas concentrations observed in obesity, diabetes, atherosclerosis and CHD are orders of magnitude lower. Further work is essential to clarify whether and how TMAO influences inflammation in these disease settings.
Vascular dysfunction and senescence
Recent studies also suggest that very high concentrations of TMAO may impair eNOS- or EDHF-dependent vascular control, and facilitate vessel remodelling and senescence(Reference Li, Chen and Gua8,Reference Li, Gua and Wu39-Reference Matsumoto, Kojima and Takayanagi41) . Elevated TMAO can increase formation of ROS that degrade NO, and may also inhibit eNOS expression, compounding vascular dysfunction (Fig. 3)(Reference Sun, Jiao and Ma19). Sun et al. (Reference Sun, Jiao and Ma19) found that 100–300 µm TMAO suppresses endothelial eNOS expression and NO release, an effect involving ROS-dependent activation of the NLRP3 inflammasome(Reference Sun, Jiao and Ma19). Additionally, Matsumoto et al. (Reference Matsumoto, Kojima and Takayanagi41) report a modest inhibitory effect of 300 µm TMAO on EDHF-mediated relaxation in the femoral (but not superior mesenteric) artery. Suggestive of some vascular influences, chronic TMAO infusion in rats to achieve a 58 µm plasma concentration did not modify blood pressure, yet prolonged the hypertensive effect of angiotensin II(Reference Ufnal, Jazwiec and Dadlez42). Whether this reflects a direct vascular influence is unclear. Supporting TMAO-dependent vascular dysfunction in disease, inhibition of TMA production with DMB reportedly preserves aortic relaxation and eNOS phosphorylation, and eliminates vascular inflammation and oxidative stress in a rat model of CKD associated with a doubling of plasma TMAO(Reference Li, Gua and Wu39). This study, remarkable in implicating a modest elevation in TMA/TMAO as the sole driver of inflammation, oxidative stress and eNOS and vascular dysregulation in CKD, requires broader confirmation and reconciliation both with evidence of other mechanistic involvement (including nuclear factor erythroid 2-related factor 2 (Nrf2) suppression, inducible NO synthase (iNOS) induction, altered parathyroid hormone and Ca2+ handling) and with the excessive TMAO concentrations required to induce such effects in vitro. There is other support for TMAO-dependent vascular damage in CKD, with vessel calcification in CKD patients correlating with TMAO concentrations above 100 µm, and evidence that 100–600 µm TMAO can promote vascular calcification and expression of bone-related and inflammatory molecules in vitro and in vivo (Reference Zhang, Li and Yang37).
Ageing is an independent risk factor for CVD and involves vascular remodelling and progressive reductions in endothelial function and NO bioavailability, coupled with increased cyclo-oxygenase (COX)-derived vasoconstrictors. Circulating TMAO increases with age in humans and rodents(Reference Li, Chen and Gua8,Reference Ke, Li and Zhao40,Reference Li, Ke and Zhan43,Reference Brunt, Gioscia-Ryan and Richey44) , and recent work indicates that inhibition of TMAO formation reduces(Reference Li, Chen and Gua8) whereas TMAO supplementation mimics or accelerates(Reference Ke, Li and Zhao40) ageing-dependent vascular dysfunction and inflammation in rodents. Pro-senescence effects were associated with exaggerated oxidative stress, suppression of sirtuin-1 and activation of p53/p21/Rb signalling(Reference Ke, Li and Zhao40). Antibiotic treatment in ageing mice also improves vascular reactivity, in association with a fall in circulating TMAO; however, the role of TMAO was not directly tested(Reference Brunt, Gioscia-Ryan and Richey44). These studies, together with evidence for TMAO-dependent brain ageing(Reference Li, Ke and Zhan43), suggest elevated TMAO could promote biological ageing, predisposing to strongly age-dependent diseases such as CHD and CKD. Indeed, Li et al. (Reference Li, Chen and Gua8) report that a moderate elevation in TMAO from about 6 to 14 µm is solely responsible for age-dependent vascular dysfunction, inflammation and eNOS depletion, with the aged vascular phenotype in senescent rats reversed by DMB treatment. This surprising observation also demands broader confirmation, and reconciliation with evidence for diverse mechanistic elements of vascular ageing. More fundamentally, TMAO concentrations required to modify viability and senescence markers in human endothelial cells(Reference Ke, Li and Zhao40) are so extreme (100 mm) as to have no biological or pathological relevance in humans or animal models.
Myocardial dysfunction
Although there are relatively few studies of the myocardial effects of TMAO, emerging evidence suggests cardiac sensitivity to concentrations that are approached or surpassed in HF and CKD. These pronounced elevations in TMAO may thus promote cardiac dysfunction, as suggested by select effects of DMB in diseased but not healthy animals(Reference Chen, Zheng and Feng10,Reference Li, Sun and Zhang45) . In addition to evidence of TMAO-dependent exaggeration of cardiac fibrosis and dysfunction in disease models(Reference Organ, Otsuka and Bhushan46), TMAO may disrupt myocyte structure, contractile function and energy metabolism, and promote oxidative stress(Reference Savi, Bocchi and Bresciani47,Reference Makrecka-Kuka, Volska and Antone48) . Inhibitory effects on contraction, relaxation and Ca2+ dynamics are evident with 20 µm TMAO(Reference Savi, Bocchi and Bresciani47), although the latter effects appeared concentration-independent in the limited range tested. However, other work indicates that 0·3–3 µm TMAO disrupts T-tubule organisation and Ca2+ handling in isolated murine cardiomyocytes(Reference Jin, Ji and Zuo49), suggesting a curious scenario in which concentrations more than 10-fold lower than circulating levels in healthy mice(Reference Chen, Zheng and Feng10) are significantly damaging. Distinct from these studies, recent work supports modest positive inotropic effects of TMAO when applied at extremely high (0·3–3 mm) levels(Reference Oakley, Vallejo and Wang50). Further work is needed to clarify the cardiac functional influences of relevant concentrations of TMAO.
Potential impacts of TMAO on cardiac energy metabolism are supported by TMAO-dependent inhibition of mitochondrial pyruvate and fatty acid oxidation, also apparent in vitro with 20 µm TMAO(Reference Makrecka-Kuka, Volska and Antone48). Another recent study suggests a threshold of about 100 µm TMAO for inhibition of oxidoreductase capacity in cultured cardiomyoblasts, although TMA appeared substantially more potent, reducing metabolic capacity at 1–10 µm (Reference Jaworska, Hering and Mosieniak51). It has also been reported that exogenously applied TMAO can destabilise atrial electrophysiology to promote fibrillation (potentially via inflammation)(Reference Yu, Meng and Huang52); however, concentrations mediating this effect are difficult to ascertain. There is also some indirect evidence that myocardial protection with physical activity may be inhibited by TMAO levels exceeding 20 µm (Reference Zhang, Meng and Yu13); however, involvement of TMAO was not tested and is inconsistent with a lack of effect of TMAO supplementation.
Apparent sensitivities to about 20 µm TMAO suggest that cardiac dysfunction could arise as a direct result of elevations observed in HF and CKD, or following acute myocardial infarction (AMI). Renal hypo-perfusion and neuroendocrine changes characteristic of HF probably contribute to the associated rise in TMAO(Reference Stubbs, House and Ocque15,Reference Missailidis, Hällqvist and Qureshi53) . Ageing, obesity, insulin resistance and diabetes(Reference Shan, Sun and Huang54) – associated with and promoting HF – may additionally contribute to increased TMAO, for example via altered flavin-containing mono-oxygenase 3 (FMO3) expression(Reference Brunt, Gioscia-Ryan and Richey44,Reference Miao, Ling and Manthena55,Reference Schugar, Shih and Warrier56) . These TMAO changes could constitute an important cardio-renal linkage, with TMAO functioning as a uraemic toxin that promotes cardiac and renal dysfunction and remodelling(Reference Ronco, Haapio and House57) (Fig. 2). Elevated TMAO does appear to worsen pressure overload-induced HF in mice(Reference Organ, Otsuka and Bhushan46), and promotes renal inflammation and fibrosis(Reference Li, Chen and Gua8,Reference Sun, Yin and Liu11) . Causal involvement in cardiac dysfunction and remodelling is supported by the observation that TMAO is elevated to about 20 µm in a rodent model of obesity, with DMB preventing diastolic dysfunction, inflammation and cardiac fibrosis independently of metabolic disruption(Reference Chen, Zheng and Feng10). The observation that myocardial impacts of dietary obesity appear entirely dependent on a 2-fold rise in TMAO (to about 20 µm) has important implications, though awaits confirmation. Li et al. (Reference Li, Sun and Zhang45) present evidence that post-infarct HF in rats may also involve an associated increase in circulating TMAO (to about 25 µm), with cardiac outcomes improved upon DMB treatment(Reference Li, Sun and Zhang45). Nonetheless, involvement of circulating TMAO in the pathogenesis of HF awaits more extensive analysis, as does its potential role as a uraemic toxin in the cardio-renal syndrome. There is contrary evidence, for example, that chronic elevations in TMAO may actually protect against myocardial dysfunction and fibrosis(Reference Huc, Drapala and Gawrys58).
Associations between trimethylamine N-oxide and CVD: cause and/or effect?
Experimental in vitro and in vivo studies to date support potentially pathogenic effects of as little as about 20 µm TMAO on vascular and cardiac function, and ≥100 µm TMAO on inflammation and other pro-atherosclerotic processes. How does this pattern of pathogenicity mesh with concentrations reported in healthy and diseased cohorts, and apparent associations between TMAO and CVD?
Measures of circulating TMAO in healthy humans (largely via stable isotope dilution and LC/MS-MS) generally range from 2 to 5 µm (Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Stubbs, House and Ocque15,Reference Mente, Chalcraft and Ak17,Reference Mafune, Iwamoto and Tsutsumi18,Reference Koeth, Wang and Levison20,Reference Ke, Li and Zhao40,Reference Missailidis, Hällqvist and Qureshi53,Reference Schugar, Shih and Warrier56,Reference Wang, Levison and Hazen59-Reference Fu, Hullar and Randolph67) , with lower estimates of 0·2–1 µm (Reference Mafune, Iwamoto and Tsutsumi18) (Table 1). Although underlying mechanisms remain obscure, circulating concentrations increase moderately (30–50 %) with body weight(Reference Schugar, Shih and Warrier56) or diabetes(Reference Schugar, Shih and Warrier56,Reference Dambrova, Latkovskis and Kuka64) , rise to about 10 µm with ageing(Reference Ke, Li and Zhao40,Reference Li, Ke and Zhan43) , and increase more profoundly in HF (>10 µm; linked to disease severity/renal dysfunction)(Reference Wang, Klipfell and Bennett2,Reference Mafune, Iwamoto and Tsutsumi18,Reference Tang, Wang and Shrestha60) and CKD (≥90 µm in end stages; also correlating with renal dysfunction)(Reference Stubbs, House and Ocque15,Reference Mafune, Iwamoto and Tsutsumi18,Reference Missailidis, Hällqvist and Qureshi53,Reference Xu, Xia and Lu68) (Table 1). Studies in rodents evidence 2- to 3-fold higher baseline concentrations than in human subjects(Reference Li, Chen and Gua8-Reference Zhang, Meng and Yu13), though some murine studies report equivalent 1–5 µm levels(Reference Ke, Li and Zhao40,Reference Brunt, Gioscia-Ryan and Richey44,Reference Organ, Otsuka and Bhushan46,Reference Dambrova, Latkovskis and Kuka64) . As in human disease, plasma TMAO increases in animal models of diet-induced obesity(Reference Chen, Zheng and Feng10,Reference Sun, Yin and Liu11,Reference Zhang, Meng and Yu13) , atherosclerosis(Reference Wang, Klipfell and Bennett2), diabetes(Reference Dambrova, Latkovskis and Kuka64) and CKD(Reference Li, Gua and Wu39), and with ageing(Reference Li, Gua and Wu39,Reference Brunt, Gioscia-Ryan and Richey44) .
Table 1. Estimates of human serum trimethylamine N-oxide (TMAO) concentrations in healthy and diseased cohorts*
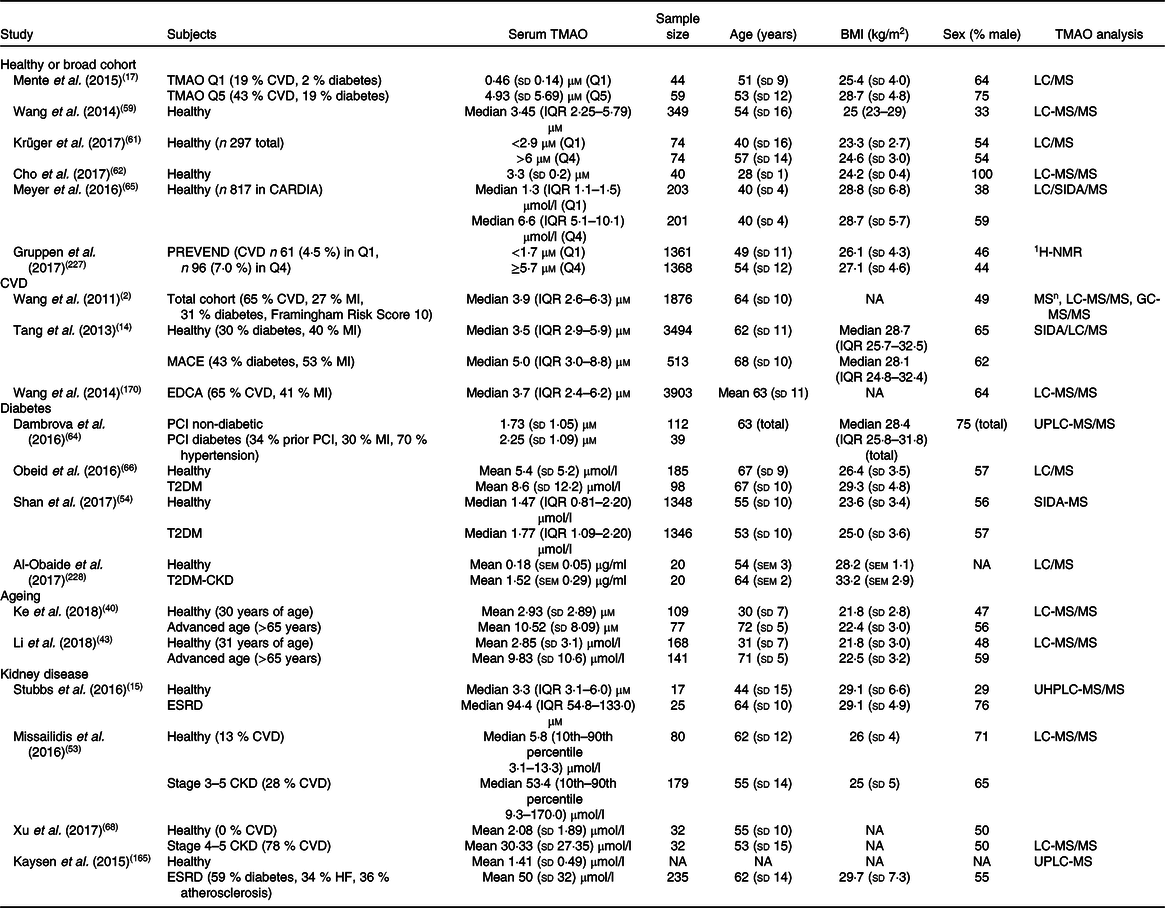
CARDIA, Coronary Artery Risk Development in Young Adults Study; CKD, complex kidney disease; EDCA, elective diagnostic coronary angiography; ESRD, end-stage renal disease; 1H-NMR, proton-NMR; IQR, interquartile range; LC, liquid chromatography; MACE, major adverse cardiovascular events; MI, myocardial infarction; MSn, multiple-stage MS; NA, not available; PCI, percutaneous coronary intervention; SIDA, stable-isotope-dilution assay; PREVEND, prevention of renal and vascular end-stage disease; T2DM, type 2 diabetes mellitus; UHPLC, ultra-high-performance liquid chromatography; UPLC, ultra-performance liquid chromatography.
* Summary of studies in human subjects detailing TMAO concentrations in healthy and pathological conditions. Values are variably expressed as: means and standard deviations, medians and IQR, or medians and 10th percentile<–>90th percentile, quartile 1 (Q1) and quartile 4 (Q4), or quintile 1 (Q1) and quintile 5 (Q5).
A number of studies link circulating TMAO with CVD(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Stubbs, House and Ocque15,Reference Missailidis, Hällqvist and Qureshi53,Reference Dambrova, Latkovskis and Kuka64) , suggesting that chronic elevations might mechanistically contribute to disease (Table 1). Several link TMAO elevations to major adverse cardiac events (MACE) in those with high cardiometabolic risk and a history of CVD(Reference Wang, Klipfell and Bennett2,Reference Mente, Chalcraft and Ak17,Reference Koeth, Wang and Levison20) , congruent with pro-atherosclerotic, vascular and myocardial effects of high TMAO in experimental models(Reference Wang, Klipfell and Bennett2,Reference Chen, Zhu and Ran7,Reference Tang, Wang and Levison14,Reference Koeth, Wang and Levison20,Reference Geng, Yang and Wang27,Reference Savi, Bocchi and Bresciani47,Reference Chistiakov, Bobryshev and Kozarov69,Reference Brown and Hazen70) . Similarly, TMAO concentrations in stable HF independently predict 5-year mortality when adjusted for traditional risk factors(Reference Tang, Wang and Fan71), and MACE either in the presence or absence of traditional CVD risks(Reference Tang, Wang and Levison14). Meta-analysis supports a concentration-dependent association between TMAO and cardiovascular risk and mortality(Reference Schiattarella, Sannino and Toscano72). These analyses collectively evidence links between TMAO concentration and CVD risk, progression and mortality in high-risk subjects.
Other evidence suggests that such associations may be limited to high-risk cohorts with existing disease or multimorbidity(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Mente, Chalcraft and Ak17,Reference Koeth, Wang and Levison20,Reference Tang, Wang and Fan71,Reference Roe, Zhang and Bhadelia73) . No association was evident between TMAO, coronary artery Ca2+ and carotid intima-media thickness in a cohort of >800 participants aged 33–55 years old without CVD(Reference Meyer, Benton and Bennett65). Association between TMAO and the metabolic syndrome in patients undergoing coronary angiography appears dependent on renal function, with no links between TMAO and history of infarction, or CHD and MACE over 8 years of follow-up(Reference Mueller, Allenspach and Othman74). These observations agree with lack of association between TMAO, plasma cholesterol and atherogenesis under normal dietary conditions in animal models(Reference Veeravalli, Karu and Scott26). Evidence that TMAO is more predictive of CVD and its outcomes in those with existing disease or co-morbidities such as diabetes(Reference Lever, George and Slow75) and CKD(Reference Tang, Wang and Kennedy76) indicates that TMAO may play a secondary or biomarker role in CVD. This is consistent with the experimental evidence that TMAO only influences disease processes under pathological conditions(Reference Geng, Yang and Wang27), without modifying vascular(Reference Li, Chen and Gua8) or cardiac phenotype(Reference Chen, Zheng and Feng10) in young healthy animals. Indeed, recent Mendelian randomisation analysis detected no relationship between genetically predicted high TMAO (or carnitine) and risk of CHD or AMI (or atrial fibrillation, stroke, type 2 diabetes mellitus or CKD), while type 2 diabetes mellitus and CKD were causally associated with elevated TMAO(Reference Jia, Dou and Gao77). These diseases may thus mechanistically drive increases in TMAO, meaning that correlations with CVD reflect a reverse causality. Critically, while a number of association studies suggest markedly increased CVD risk at ≤5 µm TMAO(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Mente, Chalcraft and Ak17-Reference Sun, Jiao and Ma19) , there is little evidence of pathological effects of TMAO at these concentrations.
Implied cardiovascular pathogenicity of trimethylamine N-oxide
The nature of reported TMAO–CVD associations raises important questions regarding causality: the TMAO concentrations linked to major increases in CVD risk or adverse outcomes are well within ranges reported in healthy cohorts (Table 1), and orders of magnitude lower than those mediating pathological effects in experimental models(Reference Seldin, Meng and Qi6,Reference Chen, Zhu and Ran7,Reference Sun, Jiao and Ma19,Reference Koeth, Wang and Levison20) . For example, it seems implausible that the TMAO–CVD association reported by Wang et al. (Reference Wang, Klipfell and Bennett2) reflects causal involvement, as it implies that a rise in TMAO from <1 to 5 µm increases CVD risk 4-fold, despite these levels falling within the range observed in healthy subjects, and no evidence of pathological responses to ≤5 µm TMAO. Similarly, the data of Tang et al. (Reference Tang, Wang and Levison14) implies that a modest 40 % rise in TMAO from 3·5 µm to 5 µm significantly promotes CVD; the association in Mente et al. (Reference Mente, Chalcraft and Ak17) suggests that a rise from <1 to 5 µm increases risk of CVD about 9-fold; and Mafune et al. (Reference Mafune, Iwamoto and Tsutsumi18) link small elevations in TMAO from <3 to 4, 5 and 6 µm to increases in diseased coronaries from 0 to 1, 2 and 3 in surgical patients with CVD(Reference Mafune, Iwamoto and Tsutsumi18). While association between TMAO and thrombosis risk suggests markedly increased thrombotic events with a rise in TMAO from 2 to 6 µm, evidence supports a 10–100 µm threshold for functional effects of TMAO in platelets(Reference Zhu, Gregory and Org21). Meta-analysis suggests a somewhat lower sensitivity of all-cause mortality, which increases about 8 % per 10 µm increment in plasma TMAO(Reference Schiattarella, Sannino and Toscano72), and highlights an unexplained disparity between TMAO levels and outcomes: mortality exhibited an unexpected 10-fold greater dependence on TMAO in the low 1–10 µm v. higher concentration ranges. In short, were these varied associations reflective of causal involvement of TMAO as an early driver of disease, normal variance observed in TMAO concentrations across healthy populations(Reference Li, Ke and Zhan43,Reference Wang, Levison and Hazen59,Reference Cho, Taesuwan and Malysheva62,Reference Meyer, Benton and Bennett65) would be predicted to profoundly increase CVD risk and mortality. However, there is little evidence for pathological effects of ≤10 µm TMAO on inflammation, atherosclerosis or thrombosis, and no pathobiological basis for markedly higher dependence of mortality on <10 µm v. >10 µm TMAO. These findings contrast evidence of a higher TMAO threshold for cardiometabolic risk approaching 10 µm (Reference Barrea, Annunziata and Muscogiuri78). Fundamentally complicating matters, the basis of the variance in TMAO in healthy populations and of elevated concentrations with CVD risks, co-morbidities and disease itself remain unclear, constraining capacity to address the question of TMAO causal involvement.
Determinants of circulating trimethylamine N-oxide in health and disease
The question of how circulating TMAO levels are regulated and increase beyond a rather broad ‘normal’ range(Reference Krüger, Merz and Rist61,Reference Wang, Bergeron and Levison79) to achieve pathological levels is fundamental to delineating the roles of TMAO in disease. Circulating TMAO, predominantly derived from bacterially generated trimethylamine (TMA) and its subsequent oxidation by hepatic FMO3, is influenced by multiple factors including age, body weight, renal clearance, metabolic and endocrine status, and diet (among others) (Fig. 2). However, these determinants only partially explain the variance in circulating TMAO in different cohorts. In subjects with dyslipidaemia, for example, TMAO concentrations correlate with age and to a limited extent BMI, yet these factors together with sex, choline concentration, renal function, and polymorphisms in FMO3 and the ABCG2 transporter may explain < 20–25 % of TMAO variance(Reference Teft, Morse and Leake80). The basis of TMAO elevations with CHD risk factors including obesity(Reference Chen, Zheng and Feng10,Reference Sun, Yin and Liu11,Reference Zhang, Meng and Yu13) , insulin resistance(Reference Fu, Hullar and Randolph67,Reference Li, Chen and Liu81) , diabetes(Reference Shan, Sun and Huang54,Reference Dambrova, Latkovskis and Kuka64,Reference Lever, George and Slow75) and the metabolic syndrome(Reference Barrea, Annunziata and Muscogiuri78) is similarly poorly defined, as is the basis of elevations in CHD. This knowledge gap, and the failure of current models to explain TMAO variance in health(Reference Krüger, Merz and Rist61) and cardiometabolic disease(Reference Teft, Morse and Leake80), point to a critical though incompletely defined role for diet–microbiota interactions and biota remodelling(Reference Wang, Bergeron and Levison79). Existence of sub-populations of biota-dependent high v. low TMAO producers(Reference Koeth, Wang and Levison20,Reference Cho, Taesuwan and Malysheva62) may also be relevant to reported links between TMAO and disease.
Dietary substrates
Excessive intake of substrates for TMA generation may contribute to elevations in circulating TMAO, influenced by gut microbiota profiles(Reference Cho, Taesuwan and Malysheva62). That said, evidence that major TMAO substrates protect against cardiometabolic disease(Reference Millard, Musani and Dibaba82) and that red meat intake is a weak CVD risk(Reference Haring, Gronroos and Nettleton83-Reference Wang, Lin and Ouyang85) appears difficult to reconcile with an important mechanistic role for TMAO in CVD. Generation of TMAO increases with intake of choline and carnitine substrates for TMA generation(Reference Rohrmann, Linseisen and Allenspach86) (Fig. 1). Increased formation occurs when sufficient choline/carnitine reaches regions of bacterial TMA production – the caecum appears to have the highest TMA generation capacity in mouse models(Reference Koeth, Wang and Levison20,Reference Koeth, Levison and Culley87) , though a recent report (albeit for a single human subject) suggests that TMA may be generated and absorbed specifically in the small intestine(Reference Stremmel, Schmidt and Schuhmann88). Such localisation, if confirmed, could increase the diet sensitivity of TMA/TMAO generation in humans v. rodents. Following gut formation and absorption, the amine is oxidised by hepatic FMO3 to TMAO (Fig. 1)(Reference Koeth, Wang and Levison20,Reference Lang, Yeung and Peter89,Reference Dolphin, Janmohamed and Smith90) . Additional forms of FMO in humans (FMO1, FMO2, FMO4, FMO5) do not appear to participate significantly in TMA oxidation(Reference Fennema, Phillips and Shephard91), whereas recent work indicates that FMO1 is responsible for about 10 % of TMA conversion in mice(Reference Veeravalli, Karu and Scott26).
Choline
Foods rich in lecithin, such as eggs, milk, liver, red meat, poultry and seafoods, are major dietary sources of choline(Reference Wang, Klipfell and Bennett2), which can be derived from fat-soluble phosphatidylcholine (the dominant source) and sphingomyelin, and water-soluble phosphocholine, glycerophosphocholine and free choline(Reference Wiedeman, Barr and Green92). The abundant choline moiety within bile presents another potential TMA substrate(Reference Roberts, Gu and Buffa22). Choline, which contains a trimethylammonium moiety, is directly converted to TMA by gut bacteria, or may be initially metabolised to γ-butyrobetaine before subsequent conversion(Reference Koeth, Levison and Culley87). There appears to be a threshold intake beyond which choline reaches the large intestine in sufficient quantities for bacterial metabolism(Reference West, Shih and Wang93,Reference Ufnal, Zadlo and Ostaszewski94) . Choline is absorbed from the small intestine via transport proteins that are 50 % saturated at 200–300 µm in mice(Reference Sheard and Zeisel95,Reference Sanford and Smyth96) . With transporter saturation, choline can reach distal sites of bacterial conversion to TMA and dimethylamine(Reference Zeisel, DaCosta and Fox97), though preliminary evidence of TMA generation within the small intestine(Reference Stremmel, Schmidt and Schuhmann88) suggests that transit to the large intestine may be unnecessary. While details of choline transport and metabolism to TMA and TMAO in humans remain to be clarified, consumption of two hard-boiled eggs (about 250 mg of choline each) together with a 250 mg radiolabelled phosphatidylcholine supplement transiently doubles plasma TMAO(Reference Tang, Wang and Levison14), similar to other studies of egg consumption(Reference Cho, Taesuwan and Malysheva62,Reference Miller, Corbin and da Costa98) (Table 2). Consistent with transience of diet-induced TMAO changes, baseline plasma TMAO in healthy subjects is not altered with 8 weeks’ consumption of six eggs per week(Reference West, Shih and Wang93). In high-fat-fed mice, supplementation with 1·2 % (w/w) of the alternative choline source sphingomyelin also increases serum TMAO, although concentrations are relatively low and insufficient to influence atherosclerosis(Reference Chung, Wang and Bursill99). Summarising studies of choline feeding: high intakes 10- to 15-fold above normal can elevate TMAO in animals(Reference Seldin, Meng and Qi6,Reference Li, Gua and Wu39,Reference Tang, Wang and Kennedy76) , as can dietary supplementation in humans(Reference Tang, Wang and Levison14,Reference Zhu, Wang and Tang100) (Table 2); however, dietary elevations appear transient, and continuous high-level intakes may be necessary to chronically elevate TMAO to pathological levels. This may involve longer-term effects of dietary substrates on TMA-generating bacteria. Further analysis of choline’s role in influencing TMAO levels and CVD is warranted, since studies indicate dietary choline may mitigate against CVD(Reference Millard, Musani and Dibaba82) or exert no effect(Reference Meyer and Shea101,Reference Nagata, Wada and Tamura102) , contrary to proposed pathogenicity of TMAO.
Table 2. Effects of diet or supplementary interventions on plasma trimethylamine N-oxide (TMAO) concentrations*
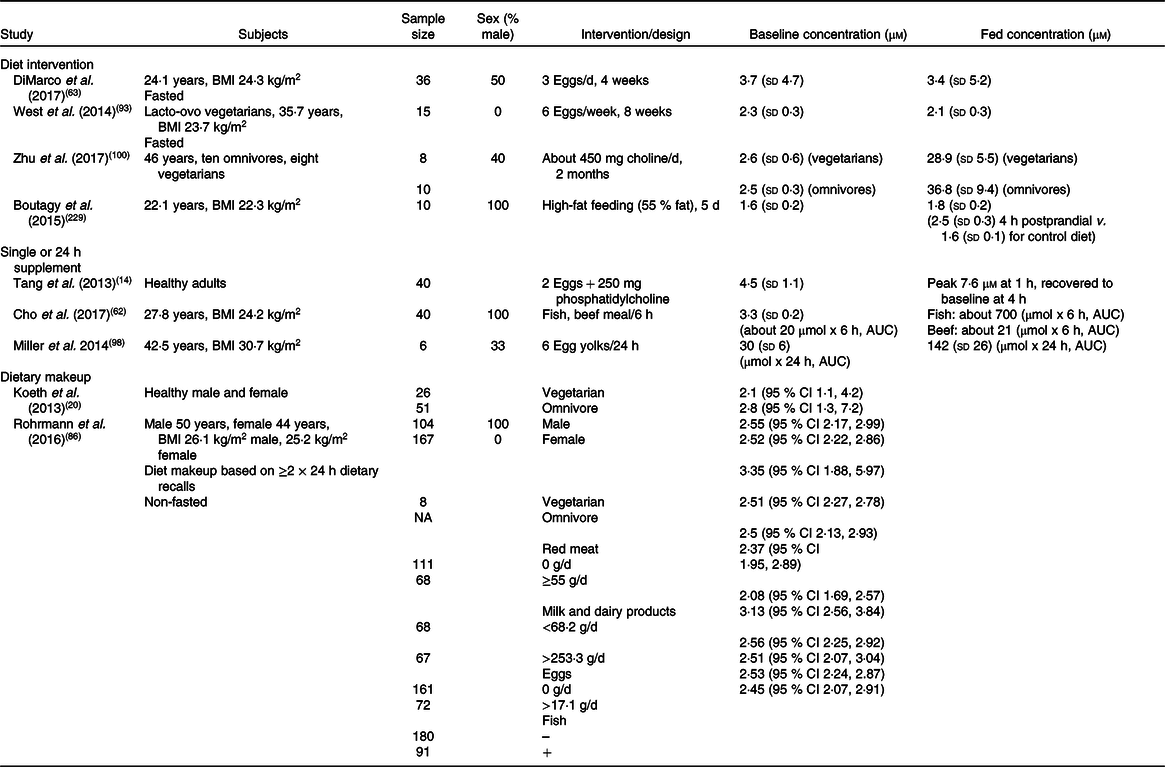
NA, not available.
* Summary of human studies examining dietary influences or effects of dietary interventions on plasma TMAO concentrations. Levels of TMAO are shown as concentration or as cumulative AUC over defined periods post-interventions, and are variably expressed as: means and standard deviations, or as means or medians and 95 % CI.
Carnitine
l-Carnitine is produced from lysine in eukaryotes, and is catabolised by prokaryotic organisms(Reference Rebouche and Seim103), the latter begin able to yield TMA and malic semialdehyde via cleavage of the backbone carbon–nitrogen bond of carnitine(Reference Meadows and Wargo104). Carnitine is an essential component of fatty acid metabolism, transporting activated long-chain fatty acyl groups into the mitochondrial matrix(Reference Hoppel105). Similar to choline, carnitine uptake from the human small intestine is not well defined and deserves further study. Mucosal carnitine uptake appears saturated with 2 g orally administered l-carnitine(Reference Harper, Elwin and Cederblad106). Saturation is also reported with 3 × 1 g doses of carnitine, significantly elevating plasma TMAO(Reference Bain, Milne and Evans107), although baseline concentrations of about 35 µm in this study are over an order of magnitude higher than widely reported(Reference Wang, Levison and Hazen59). Consumption of about 225 g of sirloin steak (about 180 mg of carnitine) transiently increases plasma TMAO concentration(Reference Koeth, Wang and Levison20). Prolonged daily supplementation of l-carnitine (1 g/d over more than 1 year) has been shown to increase median plasma TMAO by about 12-fold in a cohort of nine patients with mitochondrial disorders(Reference Vallance, Koochin and Branov108). The gut microbiota may also be able to produce γ-butryobetaine from l-carnitine metabolism(Reference Rebouche and Seim103), a metabolite that bacteria can subsequently convert to TMA(Reference Fennema, Phillips and Shephard91). Collective evidence indicates that high and chronic dietary loads of carnitine are required to elevate TMAO towards pathological concentrations (>10 µm) in the long term. Again, this may reflect substrate-driven remodelling of the gut microbiota, favouring TMAO generation (see below). Importantly, while carnitine intake increases TMAO concentrations, it reduces the risk of CVD and metabolic disorders(Reference Ussher, Lopaschuk and Arduini109), protecting against diabetes(Reference Bene, Hadzsiev and Melegh110) and the metabolic syndrome(Reference Noland, Koves and Seiler111). Meta-analysis indicates that carnitine reduces all-cause mortality, ventricular arrhythmias and angina symptoms in infarct patients(Reference DiNicolantonio, Lavie and Fares112). As for choline and seafood, the proposed pathogenic role of TMAO awaits reconciliation with these observations.
Betaine
Trimethylglycine, commonly known as betaine, provides an additional source of TMA, thus TMAO. Betaine functions predominantly as a methyl donor via the methionine cycle(Reference Craig113), and as an osmolyte(Reference Hoffmann, Brauers and Gehrmann114). The former function reduces the dietary methionine and choline required for optimal nutrition(Reference Craig113). Betaine is mainly acquired through the consumption of betaine-rich foods such as wheat bran, wheat germ and spinach(Reference Zeisel, Mar and Howe115), though irreversible oxidisation of choline to betaine within the host(Reference Weinhold and Sanders116) and gut(Reference Andresen, Kaasen and Styrvold117) represents another source. Certain gut bacteria can also convert l-carnitine into betaine(Reference Meadows and Wargo104). Betaine itself can be metabolised to TMA via multiple paths: initial conversion to choline and carnitine, as already noted; reduction to form TMA and acetate via a Stickland-type redox reaction(Reference Naumann, Hippe and Gottschalk118); or initial demethylation to dimethylglycine, which undergoes decarboxylation to form TMA(Reference Wood, Warren, Kelly and Timmis119). In terms of relationships with disease, betaine supplementation has a moderate effect on total plasma cholesterol, but not LDL, HDL or TAG(Reference Zawieja, Zawieja and Chmurzynska120), and high plasma betaine appears to have no effect on CVD risk(Reference Meyer and Shea101,Reference Guasch-Ferre, Hu and Ruiz-Canela121) while protecting against type 2 diabetes mellitus(Reference Garcia, Osté and Bennett122).
Ergothioneine
Histidine-derived ergothioneine represents another dietary precursor for TMA(Reference Muramatsu, Matsuo and Okada123). It appears ubiquitous across cells and tissues(Reference Cheah and Halliwell124), though is primarily concentrated in the liver, kidney, bone marrow and erythrocytes(Reference Melville, Horner and Lubschez125). Ergothioneine is not synthesised in animals, but is readily derived from an array of foods including mushrooms (where it is synthesised), black and red beans, oat bran and liver or kidney(Reference Ey, Schömig and Taubert126). Cellular uptake occurs via organic cation transporter 1(Reference Nakamura, Sugiura and Kobayashi127), and it may have antioxidant roles within cells(Reference Cheah and Halliwell124,Reference Halliwell, Cheah and Tang128) . The ergothionase enzyme in some gut bacteria can catalyse degradation of ergothioneine to form TMA(Reference Muramatsu, Matsuo and Okada123). To date, no studies have investigated the biological relevance of ergothioneine-dependent TMA/TMAO generation, or associations with CVD. Potential antioxidant effects of ergothioneine itself are counter to pro-oxidant and inflammatory effects of its downstream product TMAO. In terms of dietary ergothioneine sources, there is no evidence of a relationship between mushroom intake and CVD risk(Reference Lee, Yang and Giovannucci129), while there is support for increased CVD mortality v. decreased diabetes risk and cancer mortality with increased legume intake(Reference Papandreou, Becerra-Tomás and Bulló130,Reference Becerra-Tomás, Díaz-López and Rosique-Esteban131) , though the basis of these distinct disease outcomes is unknown.
Seafood
TMAO can be absorbed without gut and hepatic processing following the consumption of seafood, with plasma levels increasing within 15 min(Reference Cho, Taesuwan and Malysheva62,Reference Bain, Milne and Evans107) . Consumption of a 1·67 g dose of TMAO results in about 50 % of the molecule being absorbed unchanged(Reference Zhang, Mitchell and Smith132), while the remainder is believed to be reduced via bacterial TMAO reductase to form TMA which is subsequently absorbed and re-oxidised to TMAO(Reference Zhang, Mitchell and Smith132). Fish are rich in TMAO, owing to physiological roles in buoyancy and osmotic control(Reference Landfald, Valeur and Berstad133,Reference Withers, Morrison and Guppy134) , with the free TMAO content of seafood several-fold higher than choline/carnitine contents of red meat and eggs(Reference Landfald, Valeur and Berstad133). Populations consuming more fish exhibit high urinary TMAO concentrations(Reference Krüger, Merz and Rist61,Reference Lenz, Bright and Wilson135,Reference Dumas, Maibaum and Teague136) , and analysis of diverse foods initially revealed that only seafood substantially elevated TMAO generation and excretion in humans (>50–100-fold higher than red meat or eggs)(Reference Zhang, Mitchell and Smith132). Others confirm that eggs and red meat have relatively minor effects on postprandial plasma TMAO compared with seafood (whereas fruits reduce concentrations)(Reference Cho, Taesuwan and Malysheva62). More recently, Schmedes et al. (Reference Schmedes, Brejnrod and Aadland137) showed that a seafood meal significantly increases postprandial TMAO while a non-seafood meal does not. These potent effects of seafood intake on TMAO also await reconciliation with proposed roles of TMAO in CVD and well-established protection with seafood.
Hepatic flavin-containing mono-oxygenase 3
Circulating TMAO concentrations are dependent on TMA oxidation by hepatic FMO3, and correlate with FMO3 expression in mice and human subjects(Reference Wang, Klipfell and Bennett2).The N-oxygenation of TMA via FMO3 proceeds with a K m of 28 μm and a k cat (substrate to endproduct conversions per unit time) of >30/min, based on analysis of heterologously expressed FMO in vitro (Reference Lang, Yeung and Peter89). Thus, rapid conversion to TMAO will occur in a substrate-dependent manner in healthy individuals (TMA about 20–25 µm)(Reference Jaworska, Hering and Mosieniak51). There is evidence that hepatic FMO3 expression or activity is sensitive to sex steroids(Reference Bennett, de Aguiar Vallim and Wang28,Reference Falls, Ryu and Cao138,Reference Esposito, Varriale and D’Angelo139) , dietary factors(Reference Cashman, Xiong and Lin140) and energy intake(Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) , NO signalling(Reference Tanino, Bando and Komada143) and the transcriptional regulator CCAAT/enhancer-binding protein β(Reference Klick, Shadley and Hines144). Hepatic FMO3 expression also increases postnatally in humans, with an apparent plateau after early adulthood(Reference Koukouritaki, Simpson and Yeung145,Reference Xu, Bhatt and Yeung146) . Conversely, in female mice hepatic FMO3 transcript may decline following this phase of maturational up-regulation(Reference Fu, Csanaky and Klaassen147). While there is some debate regarding hepatic FMO expression in male rodents, an ageing-dependent increase in FMO3 protein is reported in male mice(Reference Brunt, Gioscia-Ryan and Richey44,Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) . Sex is a particularly strong determinant of FMO3 expression patterns, a linkage not strictly consistent with involvement of TMAO in CVD risk: hepatic FMO3 is substantially higher in female v. male mice and human subjects(Reference Bennett, de Aguiar Vallim and Wang28), paralleled by dimorphism in TMAO concentrations in mice(Reference Li, Chen and Gua8,Reference Bennett, de Aguiar Vallim and Wang28,Reference Ke, Li and Zhao40) and to a lesser extent humans(Reference Shimizu, Cashman and Yamazaki148), yet females enjoy protection against CVD over much of the lifespan. Pregnancy further up-regulates FMO3(Reference Hukkanen, Dempsey and Jacob149), and trimethylaminuria (a disorder in which TMA conversion to TMAO is impaired, leading to odour in sweat, urine and breath from rising TMA) is also exacerbated in women during menstruation(Reference Hukkanen, Dempsey and Jacob149). Circadian patterns of FMO3 expression may also differ markedly between sexes, a dimorphism reduced by cardioprotective energy restriction which up-regulates FMO3 in males, ‘feminising’ the transcriptional profile(Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) . However, there remains some debate regarding hepatic expression of FMO3 in male mice: there is evidence that FMO3 mRNA is below detection limits(Reference Fu, Csanaky and Klaassen147) or detectable at 0·1–10 % of the levels in females(Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) ; and that FMO3 protein is not detectable in males(Reference Chen, Yi and Zhang9,Reference Bennett, de Aguiar Vallim and Wang28) , or lowly expressed and augmented with adenoviral delivery, energy restriction or ageing(Reference Brunt, Gioscia-Ryan and Richey44,Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) . These observations raise questions regarding the utility of male murine models in assessing the role of FMO3 and TMAO in human disease.
Expression of FMO3 is modified by chronic disease and CVD risk factors, including renal dysfunction and disease(Reference Johnson, Prokopienko and West150), inflammation(Reference Zhang, Chaluvadi and Reddy151), insulin resistance and diabetes(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Shan, Sun and Huang54,Reference Miao, Ling and Manthena55) , ageing(Reference Brunt, Gioscia-Ryan and Richey44,Reference Astafev, Patel and Kondratov141,Reference Guo, Shen and Li142) , pollutants and hepatic toxins(Reference Petriello, Hoffman and Sunkara152). Elevations in TMAO itself may up-regulate FMO3 expression, with evidence of up to 5-fold induction in male mice supplemented with TMAO(Reference Ke, Li and Zhao40). Though untested (and questions remain regarding hepatic FMO3 in male mice), this suggests that a potentially detrimental positive feedback could emerge in disease settings associated with significantly elevated TMAO. Kidney dysfunction and disease not only reduce TMAO excretion but increase FMO3 expression in mice(Reference Johnson, Prokopienko and West150), further promoting elevations in TMAO. Expression of FMO3 is also increased with insulin resistance in both mice and humans(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Shan, Sun and Huang54,Reference Miao, Ling and Manthena55) , providing a basis for TMAO elevations in diabetes and a link between TMAO and CVD(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Shan, Sun and Huang54,Reference Miao, Ling and Manthena55) . Plasma TMAO in CHD patients undergoing coronary intervention can be independently linked to diabetes, together with age and BMI(Reference Dambrova, Latkovskis and Kuka64). Whether FMO3-dependent elevations in TMAO might contribute to diabetes remains to be established, though FMO3 knockdown reduces hyperglycaemia in a murine model of hepatic insulin resistance(Reference Miao, Ling and Manthena55), and circulating TMAO is predictive of insulin resistance with obesogenic feeding in macaques(Reference Li, Chen and Liu81). On the other hand, Liao et al. (Reference Liao, McManus and Hughes153) report beneficial effects of FMO3 on glucose homeostasis independent of the insulin-signalling pathway. Furthermore: hepatic TMAO concentrations decline and renal concentrations are unaltered in the db/db mouse model of diabetes(Reference Xu, Zhang and Cai154); a low-glycaemic index diet increases TMAO(Reference Barton, Navarro and Buas155) despite known benefit in obesity, diabetes and on CVD risk(Reference Augustin, Franceschi and Jenkins156); and metformin increases TMAO levels while reducing blood glucose in type 2 diabetic patients(Reference Huo, Cai and Lu157). Disruption of glycaemic control and emergence of the metabolic syndrome in mice is also independent of TMAO levels and insensitive to inhibition of TMA generation(Reference Roberts, Gu and Buffa22). Interestingly, anti-diabetic and cardioprotective resveratrol up-regulates FMO3 yet reduces TMAO levels via microbiota remodelling(Reference Chen, Yi and Zhang9,Reference Bennett, de Aguiar Vallim and Wang28) , an effect associated with reduced atherosclerosis in ApoE–/– mice(Reference Chen, Yi and Zhang9). Conversely, recent work indicates that resveratrol does not influence plasma TMAO in mice on a choline-supplemented (1 % w/w) diet(Reference Roberts, Gu and Buffa22).
Importantly, links between FMO3, TMAO and disease development are clouded by TMAO-independent effects of the enzyme. Knockdown of FMO3 improves cholesterol balance while augmenting hepatic stress and inflammation in cholesterol-fed mice independently of TMAO levels and TMA generation(Reference Warrier, Shih and Burrows158). Metabolic effects of FMO3 manipulation in LDL receptor knockout mice are TMA/TMAO independent(Reference Bennett, de Aguiar Vallim and Wang28,Reference Shih, Wang and Lee159) . Furthermore, while FMO3-dependent elevations in TMAO are suggested to promote tissue ageing(Reference Li, Chen and Gua8,Reference Ke, Li and Zhao40,Reference Li, Ke and Zhan43) , and FMO3 expression may rise with age(Reference Brunt, Gioscia-Ryan and Richey44), recent evidence indicates that increased FMO3 expression is protective, exerting anti-ageing effects that mimic those of energy restriction(Reference Guo, Shen and Li142). Thus, not only does FMO3 exert beneficial effects, but linkages between FMO3 and disease do not necessarily reflect a role for TMAO.
Cellular transport and renal excretion
Renal function is a key determinant of circulating TMAO, contributing to elevations in inter-related CKD(Reference Stubbs, House and Ocque15,Reference Mafune, Iwamoto and Tsutsumi18,Reference Missailidis, Hällqvist and Qureshi53,Reference Xu, Xia and Lu68) , CVD(Reference Wang, Klipfell and Bennett2,Reference Mafune, Iwamoto and Tsutsumi18,Reference Tang, Wang and Shrestha60) , diabetes(Reference Dambrova, Latkovskis and Kuka64) and ageing(Reference Li, Chen and Gua8,Reference Ke, Li and Zhao40,Reference Missailidis, Hällqvist and Qureshi53) . The cellular influx of TMAO may occur predominantly via organic cation transporter 2, which facilitates renal tubular TMAO uptake(Reference Teft, Morse and Leake80). Efflux occurs via ATP-binding cassette family transporters, with genetic variation in the ABCG2 transporter potentially contributing to altered TMAO concentrations in dyslipidaemic subjects(Reference Teft, Morse and Leake80). The ABCG2 gene has oestrogen and hypoxia response elements, is down-regulated by parathyroid hormone and up-regulated by aryl hydrocarbon receptors and DNA methylation, and polymorphisms influence renal excretion(Reference Kusuhara and Sugiyama160,Reference Horsey, Cox and Sarwat161) .
In healthy volunteers with normal kidney function, fractional renal clearance of TMAO exhibits a broad dynamic range(Reference Wang, Bergeron and Levison79), while patients with renal dysfunction exhibit significantly elevated plasma TMAO as a consequence of functional deficit(Reference Stubbs, House and Ocque15,Reference Missailidis, Hällqvist and Qureshi53,Reference Al-Waiz, Mitchell and Idle162,Reference Bell, Lee and Lee163) . Elevations in TMAO in CKD and HF involve changes in glomerular filtration rate and renal function(Reference Missailidis, Hällqvist and Qureshi53), with renal medullary damage with hypoperfusion additionally increasing TMAO(Reference Doucet, Dutheil and Petit164). Recent findings suggest that chronic ingestion of red meat may reduce excretion of TMAO(Reference Wang, Bergeron and Levison79). Though speculative, detrimental feedback may also emerge at the level of kidney function: as for uraemic toxins in the cardio-renal syndrome, increases in circulating TMAO due to renal hypoperfusion/injury with atherosclerosis, CHD and HF may surpass a pathological threshold to exacerbate vascular(Reference Li, Chen and Gua8,Reference Sun, Yin and Liu11) and myocardial dysfunction(Reference Savi, Bocchi and Bresciani47,Reference Makrecka-Kuka, Volska and Antone48) , renal inflammation, fibrosis and dysfunction(Reference Li, Chen and Gua8,Reference Sun, Yin and Liu11,Reference Tang, Wang and Kennedy76) (Fig. 2). Though this remains to be directly tested, elevations in TMAO predict mortality in CKD(Reference Missailidis, Hällqvist and Qureshi53,Reference Tang, Wang and Kennedy76) , and mimicking TMAO concentrations in advanced CKD and HF (80–100 µm) with either choline (1·0 % total) or TMAO (0·12 %) supplementation promotes renal fibrosis and injury in mice(Reference Tang, Wang and Kennedy76). On the other hand, others report no relationship between TMAO and mortality or cardiovascular outcomes in end-stage renal disease(Reference Kaysen, Johansen and Chertow165).
The gut microbiota and human trimethylamine N-oxide concentrations
Up to about 75 % of the variance in plasma TMAO in healthy humans appears unexplained, with meat and fish consumption estimated to account for <15 % and renal function <5 % of variance(Reference Krüger, Merz and Rist61). Age is an important factor, with substantial age-dependent elevations in TMAO in animal models and humans(Reference Li, Chen and Gua8,Reference Ke, Li and Zhao40,Reference Li, Ke and Zhan43) . Curiously, diet has been estimated to be a surprisingly weak determinant of the variance in TMAO in healthy humans(Reference Krüger, Merz and Rist61,Reference Teft, Morse and Leake80) , and analysis of the heritability of high TMAO concentrations indicates that host genetics also plays a minor role(Reference Xu, Wang and Li166), consistent with failure of gene association analysis to identify TMAO-linked loci(Reference Hartiala, Bennett and Tang167). This poor understanding of the control of TMAO levels reflects in part our incomplete (though evolving) understanding of the roles and control of gut bacterial populations in determining TMAO concentrations. Since circulating TMAO is critically dependent on gut bacteria, elevations may reflect microbiota remodelling with diet and disease. The microbiota of vegans has minimal TMAO-generation capacity(Reference Koeth, Wang and Levison20), and Cho et al. (Reference Cho, Taesuwan and Malysheva62) present evidence that distinct bacterial profiles govern TMAO generation in healthy male omnivores, identifying ‘high’ v. ‘low’ producers (42 and 58 % of volunteers, respectively). A substantial proportion of the population may thus, owing to specific microbiota profiles, be specifically susceptible to diet-dependent elevations in TMAO. Apparently paradoxical benefits of TMAO substrate intake (choline, carnitine, seafood) on cardiovascular health may also reflect dominant influences of microbiota makeup and remodelling.
The fundamental importance of the microbiota is evidenced by absence of urinary TMA in germ-free mice, and its reduction with antibiotic treatment in conventional mice(Reference al-Waiz, Mikov and Mitchell168). Effects of antibiotics on carnitine-dependent elevations in TMAO confirm microbiota dependence in humans: in female omnivores, increases in plasma TMAO with carnitine intake are abolished by antibiotics and restored after 3 weeks’ recovery for microbiota re-population(Reference Koeth, Wang and Levison20). Subsequent investigations confirm the essential role of the microbiota in humans(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14) , and differences in TMAO generation in vegans v. non-vegans further emphasise the microbiota’s importance: carnitine challenge increases plasma and urinary TMAO in omnivores but not vegans, a dimorphism eliminated by antibiotic treatment(Reference Koeth, Wang and Levison20). This suggests that intake of animal protein favours carnitine/choline-metabolising bacteria(Reference Koeth, Wang and Levison20,Reference Ferguson169) , while a non-meat diet low in these substrates and relatively enriched with betaine (a less favourable TMA substrate(Reference Wang, Tang and Buffa170,Reference Ross, Zangger and Guiraud171) ) promotes non-TMA-generating species. Recent identification of distinct bacterial profiles in high and low TMAO producers (high and low responses to dietary substrate)(Reference Cho, Taesuwan and Malysheva62), and effects of red v. white meat on bacterial carnitine v. choline metabolism(Reference Wang, Bergeron and Levison79), further confirm the importance of microbiota changes in driving TMAO elevations in humans.
Fennema et al. (Reference Fennema, Phillips and Shephard91) provide a comprehensive review and summary detailing bacterial species involved in the formation of TMA. Bacteria including Prevotella, Deferribacteres and Teneriticutes species can metabolise choline and carnitine to TMA, while Bacteroides appears less effective than Prevotella (Reference Koeth, Wang and Levison20,Reference Chistiakov, Bobryshev and Kozarov69) . A recent cross-sectional analysis of 1653 multi-ethnic participants linked circulating TMAO to the abundance of thirteen genera (six Firmicutes, three Bacteroidetes, three Protobacteria, one Fusobacteria), including Prevotella, Mitsuokella, Fusobacterium, Desulfovibrio, Bilophila and Ruminococcaceae and Lachnospiraceae family members(Reference Fu, Hullar and Randolph67). Studies indicate that omnivores possess more Firmicutes than Bacteroidetes species and a less diverse microbiota compared with vegans/vegetarians, who also have relatively lower Clostridium species(Reference Cho, Taesuwan and Malysheva62,Reference Matijasic, Obermajer and Lipoglavsek172,Reference Kabeerdoss, Devi and Mary173) . In general, meat intake appears to promote Bacteroides, Alistipes, Ruminococcus, Clostridia and Bilophila, while decreasing Bifidobacterium species(Reference Tomova, Bukovsky and Rembert174). Other work links a vegan diet to decreased Clostridium v. enrichment of Anaerostipes caccae and Lachnobacterium species in subjects with the metabolic syndrome(Reference Smits, Kootte and Levin175). Although a majority of TMA production occurs within the gut, it is also worth noting that formation can also occur in the mouth via Streptococcus sanguis metabolism of choline(Reference Chao and Zeisel176). Recent studies identify three biochemical paths for bacterial TMA production, involving the genes cutC (Reference Craciun and Balskus177), cntA (Reference Zhu, Jameson and Crosatti178) and YeaW (Reference Koeth, Levison and Culley87). Genomic assessment of TMA production potential in human bacteria, based on these pathways, identifies Firmicutes, Proteobacteria and Actinobacteria species, whereas these paths appear absent in Bacteroidetes (Reference Martinez-del Campo, Bodea and Hamer179,Reference Falony, Vieira-Silva and Raes180) . Interestingly, in terms of observed increases in circulating TMAO with ageing, Brunt et al. (Reference Brunt, Gioscia-Ryan and Richey44) report select ageing-dependent changes in mouse gut phyla (increased Proteobacteria, Verrucomicrobia, and candidate division TM7) and genera (increased Bacteroides, Akkermansia and pro-inflammatory, TMA-generating Desulfovibrio; and reductions in several Firmicutes).
Interactions between diet, microbiota makeup and TMAO generation remain to be better detailed in humans, with findings somewhat contradictory. For example, some studies indicate that vegetarian (but not vegan) diets can increase relative proportions of Prevotella, known to produce TMA(Reference Wu, Chen and Hoffmann181,Reference Ruengsomwong, Korenori and Sakamoto182) . One study found that plasma TMAO did not differ significantly between vegans and lacto-ovo-vegetarians(Reference Obeid, Awwad and Keller183), though the period the groups adhered to these diets was not provided. Recent evidence indicates that red v. white meat may selectively increase bacterial metabolism of carnitine (not choline) to TMA, while reducing renal TMAO excretion(Reference Wang, Bergeron and Levison79). Further studies are needed to clarify roles of microbiota constituents in governing TMA and thus TMAO production, and the influences of animal- and plant-derived nutrients. It is important that broader biochemical influences of TMA-generating bacteria are also detailed to better understand how the gut influences cardiovascular health – additional co-modified factors may contribute to or influence associations between TMAO and CVD. This is critical in addressing the key question of whether elevated TMAO is a biomarker of microbiota changes secondary to different CVD risk factors and co-morbidities, or is a causal factor contributing to CVD development.
Trimethylamine N-oxide: early risk biomarker and target in CVD?
Though many aspects of the regulation of TMAO accumulation and its pathogenicity remain to be resolved, assessment of evidence reviewed here leads us to speculate that modest elevations in TMAO (<10–20 µm) are unlikely to be independently pathogenic, though may serve as a biomarker of collective CVD risk with diverse factors (ageing, inactivity, hyperenergetic diet/obesity, dyslipidaemia, atherosclerosis, insulin resistance, diabetes). Each of these factors moderately elevates TMAO; however, below a 10–20 µm threshold TMAO is unlikely to mechanistically participate in CVD development. Apparent dependence of CVD risk or outcomes on these low TMAO concentrations in association studies(Reference Zhang, Mitchell and Smith132,Reference Schmedes, Brejnrod and Aadland137) is thus indirect, reflecting collective influences of multiple CVD risk factors and also protectants (for example, exercise, vegan diet), influenced by diet–microbiota interactions (Fig. 4). Targeting TMAO within this range, without attention to the underlying causal factors, may be of limited if any therapeutic value. On the other hand, elevations beyond a 10–20 µm threshold as a consequence of individual or co-morbid diseases (HF, CHD/AMI, CKD) may play an important secondary role, promoting disease progression and sequelae (Fig. 2 and Fig. 4). This is congruent with evidence that TMAO–CVD associations reflect a reverse causality (TMAO changes resulting from disease, for example, CKD or type 2 diabetes mellitus)(Reference Jia, Dou and Gao77); the restriction of TMAO–CVD associations to complex cohorts with multiple risk factors(Reference Tang, Wang and Shrestha60,Reference Tang, Wang and Fan71,Reference Lever, George and Slow75,Reference Tang, Wang and Kennedy76,Reference Troseid, Ueland and Hov184) , and evidence that such associations require one or more extant disorders(Reference Li, Chen and Gua8,Reference Li, Gua and Wu39) ; and select cardiovascular effects of inhibition of TMAO production in aged/diseased yet not young and otherwise healthy animals(Reference Li, Chen and Gua8,Reference Li, Gua and Wu39,Reference Li, Sun and Zhang45) . We thus suggest, as outlined in Fig. 4, that apparent associations between TMAO and CVD involve two phases: an early indirect phase (TMAO variance a biomarker of the balance of CVD risk factors/protectants), and a later mechanistic phase where disease-dependent elevations in TMAO may secondarily participate in cardiovascular disruption, potentially in a positive feedback manner (as speculated in Fig. 2). Nonetheless, even secondary mechanistic involvement of TMAO in CVD requires further study and confirmation.

Fig. 4. Speculated linkages between trimethylamine-N-oxide (TMAO) and CVD. Whether associations between circulating TMAO and CVD risks/outcomes reflect a causal role in disease remains unclear. Consideration of available evidence suggests indirect associations with CVD: below 10–20 µm (‘indirect’ phase), variance in TMAO reflects stimulatory influences of well-established CVD risk factors (for example, ageing, inactivity, obesity, insulin resistance, diabetes) and inhibitory influences of cardioprotectants (physical activity, vegan diets). These concentrations are insufficient to influence CVD, though may be of value as a measure of composite CVD risk. Elevations beyond this range (the ‘mechanistic’ phase) are only achieved with disease (for example, chronic kidney disease, heart failure), acute myocardial infarction in CHD patients, or co-morbid conditions (for example, type 2 diabetes mellitus (T2DM) + CHD, cardiorenal syndrome). These disease-dependent elevations in TMAO may participate in reinforcing disease development (in a potentially positive feedback manner, as speculated in Fig. 2), though this awaits confirmation. Relative effects of risk factors/disease on TMAO concentration ([TMAO]) are shown, together with approximate concentrations for TMAO-dependent pathological effects. Note the illustrative ‘CVD risk or outcomes’ y-axis range is somewhat arbitrary. EDHF, endothelium-derived hyperpolarising factor; Ins-Resist., insulin resistance; mito, mitochondrial; NOS, nitric oxide synthase; RCT, reverse cholesterol transport; SR, scavenger receptor.
Caveats to this interpretation include unknowns regarding the importance of the chronicity and temporal patterns of TMAO change, and whether TMAO may interact positively (additively, synergistically) with other risk factors to facilitate dysfunction and disease. While data are lacking, sustained low-grade elevations in TMAO could exert pathogenic effects at concentrations below thresholds for acute effects, and/or in the presence of other pro-disease factors, such as low-grade inflammation. The latter is consistent with reports that TMAO selectively exaggerates oxidised LDL-dependent but not baseline CD36 expression(Reference Geng, Yang and Wang27), and that inhibition of TMAO formation selectively inhibits inflammation, oxidative stress or cardiovascular function in aged/diseased but not young/healthy animals(Reference Seldin, Meng and Qi6,Reference Li, Chen and Gua8) . In terms of temporal patterns, one might also speculate that transient elevations or spikes in circulating TMAO concentration could be beneficial via hormesis effects, with this transient ‘stressor’ augmenting resistance to injury/stress. However, chronic low-grade elevations can exhaust such adaptive homeostasis, promoting ageing and chronic disease(Reference Lomeli, Bota and Davies185).
Manipulating trimethylamine N-oxide in CVD
As discussed above, modulation of TMAO accumulation might be beneficial in limiting disease progression and impacts in those with existing CVD, or at particularly high risk (including high TMAO producers(Reference Cho, Taesuwan and Malysheva62)). Reductions in TMAO in CVD might be achievable via modulation of both diet and the gut microbiota, though unknowns and challenges arise with each approach. Given the importance of the gut microbiota in determining TMAO production(Reference Wang, Klipfell and Bennett2,Reference Tang, Wang and Levison14,Reference Koeth, Wang and Levison20,Reference Cho, Taesuwan and Malysheva62) , its manipulation is an obvious candidate for lowering TMAO in those with CVD. Unfortunately, this is not presently feasible, as what constitutes a healthy microbiota and the roles of microbiota composition in governing TMAO concentrations are not fully understood, and our ability to selectively remodel the gut microbiota (suppressing/promoting individual species or their functionality) is not currently feasible. Antibiotics suppress TMAO production and TMAO-dependent atherosclerosis(Reference Koeth, Wang and Levison20,Reference Koeth, Levison and Culley87) ; however, this ‘shotgun’ approach is not viable given broad impacts on microbiota and host health, immunity and antibiotic resistance(Reference Brown and Hazen186). Ongoing investigations into bacterial control of TMAO generation may reveal strategies for manipulating the biota genetically, pharmacologically or via other means. For example, a recent study shows that halogen-substituted choline analogues alter the caecal microbiota and suppress elevations in TMA, TMAO and thrombosis associated with choline supplementation(Reference Roberts, Gu and Buffa22). Many drugs can modify the gut microbiota(Reference Imhann, Bonder and Vich Vila187,Reference Rogers and Aronoff188) , and a recent study indicates a quarter of 1000 drugs examined possess antibiotic-like side effects(Reference Maier, Pruteanu and Kuhn189). While this broadly supports the potential for pharmacological manipulation of the microbiota, the challenge of specificity (and untoward side effects) remains. This hurdle might be overcome with our evolving understanding of bacterial TMAO production (for example, identification of biochemical paths, and gene determinants such as cutC (Reference Craciun and Balskus177), cntA (Reference Zhu, Jameson and Crosatti178) and YeaW (Reference Koeth, Levison and Culley87)), revealing molecular targets for microbiota-directed genetic or pharmacological therapy.
Probiotics offer a low-cost and -risk approach to microbiota manipulation; however, findings are equivocal, particularly in a CVD context. Supplementation with Lactobacillus casei Shirota for >3 months (19·5 × 109 colony-forming units/d) failed to influence TMAO production in patients with the metabolic syndrome(Reference Tripolt, Leber and Triebl190), and 3-month probiotic supplementation in haemodialysis patients (9 × 1013 colony-forming units/d, including Streptococcus thermophilus, Lactobacillus acidophilus and Bifidobacteria longum) failed to influence plasma TMAO(Reference Borges, Stenvinkel and Bergman191). Meta-analyses support some cardiovascular benefits of probiotics, though outcomes are modest(Reference Khalesi, Sun and Buys192-Reference Khalesi, Bellissimo and Vandelanotte194). Unexpectedly, a small double-blind randomised controlled trial of faecal microbiota transplantation from vegans into metabolic syndrome patients successfully modified intestinal microbiota composition yet failed to alter TMAO production(Reference Smits, Kootte and Levin175). It was unclear what period of time donors adhered to vegan diets, thus whether a vegan microbiota phenotype was established, although shifts between meat and vegetarian diets induce rapid microbiota changes(Reference David, Maurice and Carmody195). While FMO3 inhibition might also appear to be a target for manipulating TMAO in patients with CVD, sufficiently effective reductions in FMO3 lead to TMA accumulation and trimethylaminuria(Reference Mitchell and Smith196). More fundamentally, FMO3 mediates diverse effects on metabolism, tissue stress and ageing independently of TMA/TMAO(Reference Guo, Shen and Li142,Reference Warrier, Shih and Burrows158) .
Dietary modification
Despite evidence that some cardioprotective diets and foods have either no effect(Reference Griffin, Djuric and Angiletta197) or increase circulating TMAO levels(Reference Zhang, Mitchell and Smith132,Reference Schmedes, Brejnrod and Aadland137) , dietary targeting of TMAO may nonetheless be of value in those with CVD. Limiting the intake of foods containing TMA precursors while increasing those favouring non-TMA-producing bacteria (for example, vegetables/fruits) or suppressing FMO3 activity (for example, indole-containing vegetables) may offer the simplest approach to reducing TMAO. Although some estimate that dietary factors are relatively weak determinants of fasting TMAO in omnivores(Reference Krüger, Merz and Rist61,Reference Teft, Morse and Leake80) , recent work supports up to 3-fold differences in circulating TMAO with red meat- v. white meat-enriched (or non-meat) diets(Reference Wang, Bergeron and Levison79). Circulating TMAO is also reportedly 2-fold higher in omnivores v. vegans(Reference Koeth, Wang and Levison20), though such differences are not always observed(Reference Rohrmann, Linseisen and Allenspach86) and warrant further study. Carnitine itself can be specifically reduced or omitted from diets; however, despite favouring TMAO accumulation, carnitine intake reduces risk of CVD and metabolic disorders(Reference Ussher, Lopaschuk and Arduini109). Meta-analysis indicates that carnitine also reduces all-cause mortality, ventricular arrhythmias and angina symptoms in infarct patients(Reference DiNicolantonio, Lavie and Fares112). Carnitine additionally induces benefits that may counter the effects of TMAO; maintaining the fermentation capacity of colonic microbiota, protecting against microbiota stressors and promoting metabolism of SCFA in association with reduced CVD risk(Reference Ghonimy, Zhang and Farouk198).
Since choline is an essential nutrient(Reference Zabell and Tang199) it may be reduced but not eliminated from the diet. However, dietary choline has been shown to have no effect(Reference Meyer and Shea101,Reference Nagata, Wada and Tamura102) or mitigate against CVD(Reference Millard, Musani and Dibaba82), though there is also some evidence that choline may increase (whereas betaine reduces) cardiometabolic risk(Reference Roe, Zhang and Bhadelia73). Studies also link choline to CVD(Reference Guasch-Ferre, Hu and Ruiz-Canela121) and MACE(Reference Wang, Tang and Buffa170) in those at high risk or with a history of CVD, and 2-month choline supplementation (about 450 mg total choline/d, equivalent to an additional 80 % of recommended intake) reportedly enhances ADP-dependent platelet aggregation in association with more than a 10-fold rise in TMAO(Reference Zhu, Wang and Tang100). Dietary sphingomyelin, which contains a choline head group, could also be reduced: it is believed to be pro-atherogenic(Reference Jiang, Paultre and Pearson200,Reference Li, Fan and Liu201) , and high circulating concentrations are linked to CVD risk(Reference Jiang, Paultre and Pearson200,Reference Nelson, Jiang and Tabas202) . However, recent findings indicate that long-term (16 weeks) sphingomyelin supplementation (1·2 % w/w) does not influence serum TMAO or atherosclerosis in ApoE–/– mice, and reduces lesion development in high-fat-fed animals(Reference Chung, Wang and Bursill99). A better understanding of choline and carnitine handling, their interactions with the gut biota, and their contributions to elevated TMA/TMAO generation is necessary in informing the manipulation of TMAO in CVD.
Shifting from an omnivorous to vegetarian diet is predicted to moderately lower baseline and postprandial TMAO concentrations(Reference Koeth, Wang and Levison20), via changes in substrate and remodelling of the microbiota. Microbiota changes with animal- v. plant-based diets are evident in as little as 5 d(Reference David, Maurice and Carmody195), and increased fruit and vegetable intake is strongly linked to protection against CVD(Reference Wang, Zheng and Yang203-Reference Glick-Bauer and Yeh207). Trials confirm that plant-based diets reduce disease progression, angina and mortality in CVD patients(Reference Esselstyn208). Nonetheless, there is limited evidence to suggest that the cardioprotective effects of vegetarian diets(Reference Wang, Ouyang and Liu205,Reference Bazzano, He and Ogden206,Reference Nagura, Iso and Watanabe209-Reference He, Nowson and MacGregor211) stem from reductions in TMAO. Relatively few investigations detail the specific impact of red meat intake on TMAO levels, an effect much less pronounced than that for seafood intake(Reference Koeth, Wang and Levison20,Reference Krüger, Merz and Rist61,Reference Cho, Taesuwan and Malysheva62) which reduces CVD risk(Reference Streppel, Ocke and Boshuizen212). Though yet to be established, long-term patterns of high animal protein consumption could exacerbate risk factors or disease-dependent elevations in TMAO, through shifts in gut bacteria, FMO3 activity and renal function(Reference Wang, Bergeron and Levison79) (for example, tubulointerstitial injury with increased dietary acid load(Reference Odermatt213,Reference van den Berg, Hospers and Navis214) ), promoting age-dependent disorders (cardiovascular and renal dysfunction, diabetes) that in turn favour further TMAO elevations (Fig. 2). Such effects might be compounded by parallel influences of metabolic state, BMI and age itself on TMAO levels. That said, although a red meat-rich diet does increase plasma TMAO(Reference Wang, Bergeron and Levison79), it is a rather weak risk for CVD(Reference Haring, Gronroos and Nettleton83-Reference Wang, Lin and Ouyang85), contrasting its strong association with other chronic diseases and the high risks attributed to small changes in TMAO(Reference Wang, Klipfell and Bennett2,Reference Mente, Chalcraft and Ak17,Reference Koeth, Wang and Levison20,Reference Tang, Wang and Fan71,Reference Wang, Tang and Buffa170) . Processed meats, on the other hand, strongly promote CVD(Reference Rohrmann and Linseisen215), yet studies demonstrating links to TMAO are lacking. Importantly, the basis of cardioprotection with a vegetarian diet or high vegetable/fruit intake is complex, and may be accounted for by non-protein components(Reference Chen, Wei and Jalili216), reductions in cholesterol and saturated fats, differing amino acid contents, and availability of dietary fibre and bioactive constituents such as isoflavones and polyphenols that counter disease processes(Reference Li, Blanco Mejia and Lytvyn217,Reference Richter, Skulas-Ray and Champagne218) . Furthermore, reducing animal protein intake (a coarse surrogate for TMAO potential) has very modest effects on CVD risk and outcomes(Reference Wang, Klipfell and Bennett2,Reference Mente, Chalcraft and Ak17,Reference Koeth, Wang and Levison20,Reference Tang, Wang and Fan71,Reference Haring, Gronroos and Nettleton83-Reference Wang, Lin and Ouyang85,Reference Wang, Tang and Buffa170) and benefit is difficult to attribute specifically to protein source(Reference Richter, Skulas-Ray and Champagne218). Other studies indicate that high protein intake reduces cardiometabolic risk, BMI and blood pressure, effects more pronounced for plant yet still evident with animal protein(Reference Berryman, Agarwal and Lieberman219,Reference Mehrabani, Asemi and Najafian220) . There is also evidence that the link between high animal protein intake and mortality requires co-existence of one or more additional risk factors(Reference Song, Fung and Hu221), consistent with evidence that links between TMAO and disease require the existence of one or more chronic disorders(Reference Li, Chen and Gua8,Reference Li, Gua and Wu39) . Finally, as seafood is cardioprotective and recommended for CVD risk reduction, omission from diets may be detrimental. Well-established protection with seafood is itself difficult to reconcile with an important role for TMAO as a diet-related risk factor, since seafood increases TMAO(Reference Krüger, Merz and Rist61,Reference Lenz, Bright and Wilson135,Reference Dumas, Maibaum and Teague136) well beyond levels achieved with other foods(Reference Zhang, Mitchell and Smith132,Reference Schmedes, Brejnrod and Aadland137) . Although a recent experimental study suggests that fish intake might promote atherosclerosis in rodents(Reference Yazdekhasti, Brandsch and Schmidt222), this controversial finding contrasts anti-atherosclerotic and cardioprotective effects of seafood in humans and awaits confirmation.
Specific foods do have potential to modulate TMAO: grapefruit juice and indole-containing vegetables can decrease FMO3 activity and alter TMAO metabolism(Reference Bain, Fornasini and Evans223). Recently, grape pomace polyphenol supplementation has been shown to decrease serum TMAO in a cohort of healthy(Reference Annunziata, Maisto and Schisano224) and overweight/obese patients(Reference Annunziata, Maisto and Schisano225). Whereas Brussels sprouts have the highest choline content among vegetables, increased intake for 3 weeks significantly decreases circulating TMAO, an effect attributed to decreased FMO3 activity without generating trimethylaminuria(Reference Cashman, Xiong and Lin140,Reference Busby, Fischer and da Costa226) . Other foods associated with a cardioprotective Mediterranean diet appear to be significant sources of endogenous DMB, including some balsamic vinegars, red wines, cold-pressed extra-virgin olive oils and grape seed oils, with levels as high as 25 mm (Reference Wang, Roberts and Buffa23).
Conclusions and future directions
CVD remains the leading cause of morbidity and mortality globally, placing an enormous burden on health systems, economies and the individuals directly and indirectly affected. A proposed role for the microbiota-dependent amine TMAO as a new and modifiable determinant of CVD has thus generated much excitement. However, much remains to be clarified regarding the control of TMAO concentrations and its potential involvement in disease. Variations in human TMAO concentrations remain largely unexplained, and whether pathologically relevant elevations arise independently of other disorders is unclear. Although increased concentrations of TMAO can promote inflammation, atherosclerosis, vascular and cardiac dysfunction and remodelling, levels inducing these effects may only be achieved in HF or CKD, or potentially CHD with co-morbid conditions (or AMI). In these select settings TMAO could play a secondary reinforcing role (Fig. 2 and Fig. 4), though even this mechanistic contribution awaits confirmation. A mechanistic role for TMAO in the development of CVD also requires reconciliation with the protective effects of its dietary precursors (particularly seafood and carnitine), and the low CVD risk associated with red meat intake. Future studies should more directly test the mechanistic relevance of TMAO in CVD, clarify the effects of chronic low-grade changes in TMAO, and test whether speculative positive feedbacks (as outlined in Fig. 2) might lead to progressive elevations in TMAO and dysfunction in CVD. This model is untested, though informed by knowledge that putative effects of TMAO (for example, inflammation, renal dysfunction and hypoperfusion) can further enhance TMAO accumulation, and observations that TMAO and renal dysfunction may up-regulate FMO3(Reference Ke, Li and Zhao40), for example. Importantly, even a secondary reinforcing role supports both the utility of TMAO as a biomarker of CVD risk, and as a therapeutic target in high-risk subjects with multiple co-morbidities or extant CVD. How to specifically reduce TMAO without potentially detrimental effects nonetheless poses a challenge. Enhanced understanding of the specific roles of bacteria in governing TMAO concentrations and how they respond to dietary modulation, together with factors influencing FMO3 activity and other determinants of TMAO concentration, is necessary before potential benefits of TMAO manipulation might be realised in select disease settings.
Acknowledgements
Fig. 1 and Fig. 2 were created using illustrations sourced from Smart Servier Medical Art.
The authors were supported by research funds from Griffith University. S. N. is supported by the Abedian Foundation Top Up Scholarship (AbedianTOP).
S. N. and J. P. H. wrote the first draft. A. J. C., E. F. D. T. and J. N. P. revised the manuscript and approved the final draft.
The authors have no relevant interests to declare.






