


Introduction
Hydrogen is a promising energy vector, a valuable industrial building block, and a key reagent for the thermochemical conversion of CO2 into fuels and chemicals.[ Reference Dunn 1 , Reference Holladay, Hu, King and Wang 2 ] The challenge is to produce it sustainably and cost-effectively. To date, renewable means of H2 production such as water electrolysis have failed to achieve cost-parity with fossil fuel-based steam methane reforming and account for <5% of global H2 production.[ Reference Simbolotti 3 , Reference Rostrup-Nielsen and Rostrup-Nielsen 4 ] While the cost of renewable electricity has historically comprised a large fraction of the overall cost of clean H2 production, it is falling rapidly, making other costs increasingly more relevant.[ Reference Satyapal 5 ] Chief among these is the high cost of the precious metal catalysts used in conventional proton-exchange water electrolyzers.[ Reference He, Desai, Brown and Bollepalli 6 , Reference Liu, Lu, Luo, Sun and Asiri 7 ] Although the high performance of platinum and other rare d-metals justify their use for small-scale H2 production, they are too scarce to satisfy the world's need for clean H2. To the extent that water electrolysis could ever be regarded as a viable means of large-scale H2 production, there is therefore significant global interest in the development of effective non-precious hydrogen evolution reaction (HER) electrocatalysts.
Some of the most promising non-precious HER catalysts are functional oxides of Ni and Ni-based alloys, which tend to be stable only in alkaline environments. One of the important features of electrocatalysis in base is the large number of oxidation states available to most metals.[ Reference Danilovic, Subbaraman, Strmcnik, Chang, Paulikas, Stamenkovic and Markovic 8 , Reference Quaino, Juarez, Santos and Schmickler 9 ] Sabatier style volcano plots, which have been successful in understanding and designing high-performance HER catalysts in acidic environments,[ Reference Kibsgaard, Chen, Reinecke and Jaramillo 10 – Reference Nørskov, Bligaard, Logadottir, Kitchin, Chen, Pandelov and Stimming 12 ] are therefore less developed for alkaline systems because the surface structure and correlated binding energy of hydrogen are difficult to observe or predict. While the multitude of possible phases complicates analyses of these systems, the coexistence of different oxidation states is central to the catalytic mechanism of functional oxide materials. Cooperation between Ni and various oxide phases, such as NiO, NiO(OH), and Ni(OH)2, and even between sulfides of Co and Mo have been proposed as integral to the performance of various alkaline HER systems.[ Reference Danilovic, Subbaraman, Strmcnik, Chang, Paulikas, Stamenkovic and Markovic 8 , Reference Staszak-Jirkovský, Malliakas, Lopes, Danilovic, Kota, Chang, Genorio, Strmcnik, Stamenkovic, Kanatzidis and Markovic 13 – Reference Wang, Lee, Deng, Lu, Hsu, Liu, Lin and Cui 15 ] The plurality of implicated phases points to a bifunctional mechanism, in which certain sites help stabilize water as a proton is adsorbed onto the surface [Eq. (1)], and different sites catalyze either a Tafel [Eq. (2)] or Heyrovsky step [Eq. (3)] for H2 gas evolution.
 $${\rm H}_2{\rm O} + e^ - \to \,{\rm H}_{{\rm ads}} + {\rm O}{\rm H}^ -, $$
$${\rm H}_2{\rm O} + e^ - \to \,{\rm H}_{{\rm ads}} + {\rm O}{\rm H}^ -, $$
 $$2{\rm H}_{{\rm ads}}\, \to \, {\rm H}_2,$$
$$2{\rm H}_{{\rm ads}}\, \to \, {\rm H}_2,$$
 $${\rm H}_2{\rm O} + {\rm H}_{{\rm ads}} + {\rm e}^ - \to \,{\rm H}_2 + {\rm O}{\rm H}^ -. $$
$${\rm H}_2{\rm O} + {\rm H}_{{\rm ads}} + {\rm e}^ - \to \,{\rm H}_2 + {\rm O}{\rm H}^ -. $$
Nickel–molybdenum (Ni–Mo) alloys in particular have emerged as the best-performing non-precious metal catalysts for HER in base, with onset potentials competitive with electrodeposited Pt.[ Reference McKone, Warren, Bierman, Boettcher, Brunschwig, Lewis and Gray 16 – Reference Raj and Venkatesan 21 ] However, while both co-electrodeposited and co-synthesized Ni–Mo alloys have been subjects of extensive study, the nature of the active site or sites and the mechanism of enhancement have remained unclear. A key problem is that the ill-defined electrodeposited or multilayered materials commonly used in studies of Ni–Mo introduce a degree of variability that is hard to account for. For codeposited systems, the Ni–Mo ratio strongly affects not only the nature of the surface but also the textural properties of the electrode. Mo dissolution and redeposition also clearly play a role in both the initial enhancement and slow-degradation of Ni–Mo alloys[ Reference Raj 22 – Reference Brossard 24 ] but are difficult to disentangle from the fabrication process for codeposited electrodes. To begin to elucidate the synergistic effect in Ni–Mo alloys, we therefore sought to create a system with well-defined Ni–Mo interface geometries. By depositing pre-formed monodisperse Ni nanocrystals (NCs) on both Mo and inert metal substrates, we are able to disentangle fabrication and catalysis effects to better probe the nature of the active site(s).
Experimental
The 12 nm (large) and 7 nm (medium) Ni NCs were prepared as described previously,[ Reference Cargnello, Doan-Nguyen, Gordon, Diaz, Stach, Gorte, Fornasiero and Murray 25 ] while 5 nm (small) NCs were synthesized by a slightly modified procedure using 30 equivalents of trioctylphosphine (TOP) and 60 equivalents of oleylamine (OLAM). Briefly, Ni(acac)2 (Sigma-Aldrich, 97%) and OLAM (Sigma-Aldrich, 70%) were dissolved in 15 mL of benzyl ether (Acros Organics, 99%) and evacuated to 1 mmHg at room temperature for 15 min. Next, an appropriate amount of TOP was added and the mixture was further evacuated at 100 °C for 30 min. The reaction mixture was then put under a static N2 atmosphere, and heated to 230 °C for 15 min. Finally, the mixture was cooled to room temperature and the NCs were purified by a repeated process (three times) of precipitation from isopropanol, centrifugation, and redispersal in hexanes. The as-synthesized NCs were characterized using a FEI Tecnai G2 F20 X-TWIN transmission electron microscope (TEM).
Sub-monolayer nickel (Ni) nanoparticle films were prepared by dropcasting a dilute (30 µg/mL) solution of Ni NCs in hexanes onto clean 1.5 × 1.5 cm2 Mo (Aldrich) and titanium (Ti) (GalliumSource) foil substrates. Evaporation of the hexanes was slowed by covering the drying substrate. A small amount of OLAM (50 µL in 20 mL hexanes) was added to the NC solution to prevent aggregation of the particles prior to dropcasting. Finally, the films underwent a fast thermal treatment, 5 min at 500 °C, to remove ligands.[ Reference Cargnello, Chen, Diroll, Doan-Nguyen, Gorte and Murray 26 ] The resulting NC films were analyzed using a FEI Magellan 400 XHR scanning electron microscope (SEM).
Ni NC films were characterized at room temperature in a three-electrode compression cell with a constant geometric electrode area of 0.95 cm2. Platinized titanium (Open Water Power) was used as a counter electrode. Voltage was monitored relative an Ag/AgCl (4 M KCl, Aldrich) reference electrode using a potentiostat (BioLogic SP-240). The 2 M KOH electrolyte was continuously purged with a vigorous flow of H2 gas (99.999%) to maintain a constant H2 activity. Chronoamperometric techniques were used to assess the catalytic performance of the different NC films, as cyclic voltammograms may lead to cycles of Mo dissolution and redeposition in sweeps to more cathodic potentials.[ Reference Takeno 27 ]
Results and Discussion
Figure 1(a)–(c) show representative TEM images of the initial Ni NC precursors. The Ni NCs are highly uniform with average particle diameters of 5, 7, and 12 nm (small, medium, and large) and 1σ particle size distributions of 11%, 13%, and 16%. While the large Ni NCs show a slightly wider particle size distribution, it is still outside the size window of the other two samples. Hence, there is minimal if any overlap in particle size between different samples, lending each film the distinct interface geometry which is crucial for elucidating catalytic interactions. Taking advantage of the colloidal nature of the Ni NC precursors, a drop-casting technique is employed to deposit them onto substrates with different catalytic properties, here Ti and Mo thin foils, with identical geometric areas. Because the same three Ni NC precursors are utilized for both substrates with identical metal loading, this library explores fundamental interactions by comparing samples where one single parameter is varied each time, whether it is particle size or the chemical nature of the substrate.

Figure 1. (a–c) TEM of Ni NCs. Insets at the bottom of each image show histograms of NC size, binned identically on an axis spanning 0–20 nm. (d–f) SEM of NCs as deposited on Mo. (g–i) SEM of NCs as deposited on Ti.
Representative SEM images of the NC films as cast onto both Mo [Fig. 1(d)–(f)] and Ti [Fig. 1(g)–(i)] substrates after thermal treatment to remove NC organic ligands confirm that the morphology of individual particles has been essentially maintained. However, two contrasting elements between the substrates are immediately clear from the images: (i) Ni NCs are much better dispersed on the Mo substrate compared with the Ti, although they are still present as individual particles in both cases; (ii) NC diameter for all three sizes appear to be much larger on Mo than on Ti substrates or in the as-synthesized state on TEM grids. In particular, the magnitude of the diameter increase (>100%) is too large to be due to oxidation of the particles alone, although this effect would certainly contribute. Based on the lattice parameters and crystal structures of Ni and NiO,[ Reference Cardarelli 28 ] total oxidation of the particles should increase the large NC apparent diameters by maximum 20%. Sintering also fails to explain the observed size increase; as shown in Fig. 2(a), multiple layers of particles on a glassy carbon substrate appear the same size as identical particles viewed in TEM. This observation is consistent with previous work showing that fast annealing to remove ligands does not cause sintering in the NC layers.[ Reference Cargnello, Doan-Nguyen, Gordon, Diaz, Stach, Gorte, Fornasiero and Murray 25 ] The different point spread functions of the SEM and TEM optics were also considered as potential explanations for the observed size disparity, but the ligand-coated NCs on TEM grids have approximately the same apparent size when viewed in either instrument. Ultimately, by tilting the substrate inside the SEM and viewing the particles at a 30° angle, it was determined that the apparent size increase of the Ni NCs on Mo supports is a result of wetting of the NCs on the substrates. As shown in Fig. 2(b), the NCs appear to have strongly favorable interaction with the Mo substrate, and wet the surface with a low contact angle (possibly <45°). These observations point to strong interactions between the Ni particles and the Mo foil.

Figure 2. (a) A high mass-loading yields multiple layers of nominally 5 nm NCs on a glassy carbon substrate. These NCs do not sinter, and appear approximately the same size as identical particles viewed in the TEM [Fig. 1(b)]. (b) A low mass-loading of nominally 7 nm Ni NCs wetting a Mo substrate, viewed at an SEM stage tilt angle of 60°.
Figure 3 shows the electrocatalytic performance of medium-sized Ni NC films on both Ti and Mo substrates and that of the respective bare substrates at −0.27 V versus the reversible hydrogen electrode (RHE). Gas flow measurements confirmed that H2 production occurs at nearly 100% Faradaic efficiency. The activity of both bare substrates is very low, with Mo still showing eight times higher current than Ti. For films of medium-sized Ni NCs, the current density increases in both cases but by drastically different amounts. While the activity of the Ni NC on Ti is still modest and only barely higher than that of the Mo foil, a large boost in current density is seen for the Ni NCs deposited on the Mo substrate. The increase in current density for the Ni/Mo sample greatly exceeds the sum in activity of bare Mo substrate and the Ni/Ti sample, implying a synergistic effect between Ni and Mo.

Figure 3. Electrocatalytic performance of bare Ti and Mo substrates, as well as medium-sized Ni NC films on both substrates at −0.27 V versus the regular hydrogen electrode (RHE). Current densities are normalized to electrochemical surface areas established using cyclic voltammograms at different scan rates, as described by many authors.[ Reference Li, Ciston and Kanan 30 ]
SEM characterization of samples after electrocatalytic activity shows distinct Ni and Mo phases, suggesting that a bulk alloy was not formed. Furthermore, the timescale of the enhancement is too short to be consistent with a Mo dissolution and redeposition process, which has been seen to produce an enhancement arising from the formation of Ni–Mo alloys, but only at larger timescales.[ Reference Brossard 24 ] Both of these observations point to an enhancement mechanism that is unrelated to the formation of a bulk alloy.
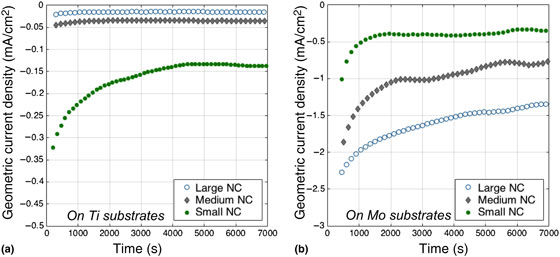
In order to further explore the physical origin of the observed synergy, different sizes of Ni NCs supported onto Ti and Mo were tested under the same conditions for HER in base. This set of measurements helps clarify whether surface area, interfacial area or perimeter length are responsible for the observed activity in accordance to well-established models.[ Reference Kibsgaard, Chen, Reinecke and Jaramillo 10 , Reference Cargnello, Doan-Nguyen, Gordon, Diaz, Stach, Gorte, Fornasiero and Murray 25 , Reference Shekhar, Wang, Lee, Williams, Kim, Stach, Miller, Delgass and Ribeiro 29 ] The data for Ni–Ti samples [Fig. 4(a)] show that small NCs deliver higher current densities compared with medium and large NCs, in this order. The trend is easy to reconcile with exposed Ni surface area at each metal loading, which causes smaller particles to deliver higher rates. Surprisingly, the data for Ni–Mo show the opposite trend. Large NCs on Mo are more active than medium and small NCs, as reported in Fig. 4(a). These data demonstrate that contact area, perimeter length or interface area between Ni and Mo are not the root causes for the observed synergy, which would otherwise still favor small NCs.[ Reference Cargnello, Doan-Nguyen, Gordon, Diaz, Stach, Gorte, Fornasiero and Murray 25 ] Rather, these observations suggest a mechanism that relies on electronic interactions between the Ni NCs and the Mo substrate. Partial reduction of the Ni NCs by Mo to a more metallic state, leading to the coexistence of multiple Ni oxidation states on the same particle, could cause the observed inverse activity trend. If smaller particles are too susceptible to this reducing influence, they may not be able to achieve the mix of Ni oxidation states associated with high activity, explaining the unexpected size dependence in Fig. 4. Larger particles, instead, would contain both domains that are directly influenced by the underlying Mo substrate, and other domains that are essentially only influenced by the reaction conditions. This mechanistic understanding is consistent with previous findings in which NiO on Ni is more active than Ni or NiO alone.[ Reference Gong, Zhou, Tsai, Zhou, Guan, Lin, Zhang, Hu, Wang, Yang, Pennycook, Hwang and Dai 14 ] While further analysis of the particle composition or the reaction conditions in situ would be required to reflect dispositively on a possible bifunctional mechanism, the mildness of the heat treatment and the inverse dependence on the Ni–Mo interfacial area strongly suggest that the observed enhancement is not related to alloy formation, but is rather an effect of bulk Mo on the oxidation state of the Ni NCs.

Figure 4. Electrocatalytic performance of sub-monolayer films of small, medium, and large Ni NC on both (a) Ti and (b) Mo substrates at −0.27 V versus RHE. The different size dependence on Ti versus Mo substrates is likely due to the stabilization of reduced Ni by Mo, which allows only the larger particles to maintain both the regions of reduced and oxidized Ni required for a bifunctional catalytic mechanism.[ Reference Danilovic, Subbaraman, Strmcnik, Chang, Paulikas, Stamenkovic and Markovic 8 , Reference Gong, Zhou, Tsai, Zhou, Guan, Lin, Zhang, Hu, Wang, Yang, Pennycook, Hwang and Dai 14 ] Normalizing by Ni surface area exaggerates both trends, while normalizing by electrochemical surface area renders the absolute activity consistent with Fig. 3, as shown in the Supplemental Information.
Conclusions
Molybdenum substrates enhance the HER activity of Ni NC electrocatalysts. There is an unexpected size dependence, in which larger particles are more active than smaller particles on a mass basis. This inverse dependence indicates that the enhancement is not due to an alloy phase at the Ni–Mo interface, but is rather an electronic effect of bulk Mo on nano Ni domains. In this case, partial reduction of Ni could allow for the coexistence of multiple oxidation states on larger particles, leading to higher activity via a bifunctional reaction mechanism.
Supplementary material
The supplementary material for this article can be found at http://dx.doi.org/10.1557/mrc.2016.27
Acknowledgments
J. A. S. acknowledges support of the Stanford University Center for Interface Science and Catalysis (SUNCAT) seed funding from the Dean of Engineering and the Dean of Research at Stanford University. I. S. M. was supported by the Department of Defense through the National Defense Science & Engineering Graduate Fellowship Program, and by the Fannie and John Hertz Foundation through a Hertz Foundation Fellowship. M. C. acknowledges support from the School of Engineering at Stanford University and from a Terman Faculty Fellowship.





