


1. Introduction
Familial hypertrophic cardiomyopathy (FHCM; OMIM 191045) is an autosomal dominant disorder characterized by hypertrophy of the left ventricle and the interventricular septum (without dilatation of the ventricular chamber), and cardiac myocyte disarray (Chung et al., Reference Chung, Tsoutsman and Semsarian2003). Hypertrophic cardiomyopathy (HCM) is a leading cause of sudden cardiac death (SCD), particularly in young adults and competitive athletes (Maron et al., Reference Maron, Estes, Maron, Almquist, Link and Udelson2003), and is present in approximately two per 1000 people (Maron, Reference Maron2002). The disease is phenotypically heterogeneous. Some individuals remain asymptomatic throughout life, and others develop progressive symptoms with or without heart failure, or experience SCD. At least 13 genes have been identified to be responsible for FHCM, all encoding components of the contractile apparatus of the cardiac myocyte (Marian & Roberts, Reference Marian and Roberts2001). Interestingly, the prognosis of patients with FHCM, caused by different gene mutations or different mutations in an identical gene, differs greatly (Taylor et al., Reference Taylor, Carniel and Mestroni2004).
Troponin T (TnT) is one of the three subunits that form the troponin complex, which together with tropomyosin, is responsible for the regulation of striated muscle contraction. TnT now appears to be the key regulator of the sarcomere, which binds the two other subunits to tropomyosin and itself binds the whole protein complex to the actin thin filament (Li et al., Reference Li, Wang and Sykes2004). In fact, the TnT gene is the third most common gene responsible for FHCM, accounting for approximately 15–20% of all cases (Knollmann & Potter, Reference Knollmann and Potter2001). Patients with FHCM caused by the TnT gene mutation have clinically variable symptoms with two general features: (i) a high risk of SCD and (ii) varying disease penetrance with only moderate hypertrophy (Gomes et al., Reference Gomes, Barnes, Harada and Potter2004).
In this study, we recruited a large multi-generational Chinese family affected by HCM. Through genome-wide screening and linkage analysis, the disease locus for this family affected by FHCM was mapped to chromosome 1q32 near the marker, D1S249. Furthermore, a novel missense mutation, Ile90Met, within the cardiac TnT (cTnT) gene was identified and confirmed to be responsible for FHCM in this family.
2. Subjects and methods
(i) Subjects and clinical evaluation
A five-generation family that consisted of 89 individuals with FHCM was identified in the Zejiang Province, China. The study conformed to the guidelines involving human research as stated in the Declaration of Helsinki. There were 12 affected members (six males and six females). Four patients died, two of whom had SCD. After informed, written consent was obtained in accordance with the study protocol approved by the local Ethics Committee, 67 family members received careful clinical examinations, and electrocardiographic and echocardiographic assessments by experienced physicians in the Sixth People's Hospital of Shanghai. Blood was drawn from family members for genetic analyses. The maximal thickness of the left ventricular wall was measured in diastole in the region of greatest hypertrophy. The diagnosis of HCM was based on the demonstration of a maximal left ventricular wall thickness of at least 13 mm in the absence of other causes of ventricular hypertrophy (Taylor et al., Reference Taylor, Carniel and Mestroni2004). Subjects with a maximal left ventricular wall thickness of <13 mm and without electrocardiographic features suggestive of HCM, but who were later shown to carry the gene defect, were classified as having a non-penetrant mutation.
(ii) Genotyping and linkage analyses
DNA was obtained from peripheral blood by standard proteinase K digestion and phenol–chloroform extraction. Genome-wide screening was performed using 382 fluorescent-labelled polymorphic micro-satellite markers from autosomes, at a resolution of approximately 10 cM (Linkage Mapping Set, version 2.5; PerkinElmer, Foster City, CA, USA). The markers were amplified by a PCR under the following conditions: 50 ng of genomic DNA, 2 pmol of each primer, 0·2 μl of dNTP (10 mM each), 1 μl of 10×buffer, 0·6 μl of MgCl2 (25 mM) and 0·4 unit of HotStar Taq DNA polymerase (Qiagen, Santa Clarita, CA, USA), in a final volume of 10 μl. Six to eight primer pairs were multiplexed in the amplification reaction. Samples were incubated in a PTC-225 DNA Engine Tetrad (MJ Research, Waltham, MA, USA) for 15 min at 95°C for pre-denaturing, followed by 9 cycles of 30 s at 94°C, 30 s at 55°C and 2 min at 72°C and then 29 cycles of 30 s at 94°C, 30 s at 52°C and 2 min at 72°C; a final extension at 72°C for 15 min was performed. Amplification products were appropriately pooled into prescribed panels, diluted and denatured for 5 min at 95°C and then ice-bathed for 2 min. Subsequently, the products were run in an automated DNA sequencer (ABI Prism 3700; Applied Biosystems, Foster City, CA, USA). The size of the allele was determined on the basis of an internal size standard (Genescan-500 LIZ; PerkinElmer) in each lane.
Data were analysed using GeneScan 3.7NT and Genotyper 3.7NT software (PerkinElmer). Statistical analysis was performed based on an autosomal dominant disease. Two-point LOD scores were calculated using the MLINK program from the Linkage software package (version 5.2, available at http://linkage.rockefeller.edu/soft/fastlink). Haplotype analysis was performed with Cyrillic software (version 2.0, available at http://linkage.rockefeller.edu/soft/fastlink). Calculations of LOD scores were performed with the assumption of a disease penetrance of 0·95 and a phenocopy rate of 0·01.
For fine mapping, two additional micro-satellite markers were selected from the genetic map: D1S2622 and D1S2655. The 5′ terminus of each primer set was modified with fluorescent carboxyfluorescein (FAM), a material producing red fluorescence. Primer information was obtained from the Genome Database (http://www.gdb.org). Two-point LOD scores were also calculated.
(iii) Mutations analysis of the cTnT gene
The mutations in all 14 exons and the flanking intronic sequences of the cTnT gene were screened from members of this family by directed sequencing. Using Soft Primer Express 2.0 (PerkinElmer), primers were designed to amplify all exons and flanking intronic splicing regions of TnT from genomic DNA (the sequences of all primers used in this study are available upon request). Screening for mutations was performed in two affected and two unaffected individuals. PCR was carried out using Taq DNA polymerase and the products were sequenced directly with a dye-terminator cycle-sequencing system by an ABI Prism 3700 DNA sequencer after purification by exonuclease I (Epicentre, Madison, WI, USA) and shrimp alkaline phosphatase (USB, Cleveland, OH, USA). The resulting sequences were compared with the corresponding wild-type sequences using AutoAssembler software (version 2.0; PerkinElmer). When sequence variants were detected, the exon was amplified from the genomic DNA extracted from the other individuals in the family affected with HCM to determine whether the base variant was specific to the patients.
3. Results
One large multi-generational family with autosomal dominant HCM was characterized (Table 1 and Fig. 1). Twelve family members had HCM without conduction-system disease, skeletal-muscle dysfunction, or other phenotypes. Analysis by echocardiography showed that all individuals had a maximal left ventricular wall thickness of at least 13 mm. The distribution of this disease was equal in males and females. The mean age at diagnosis was 48·2±15·2 years (mean±SD). The mean maximal left ventricular wall thickness was 21·6±3·7 mm. Progressive ventricular dysfunction caused congestive heart failure in subject II-2. SCD occurred in subjects II-8 and IV-18, at 37 and 19 years of age, respectively (Table 1).
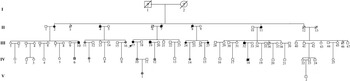
Fig. 1. A pedigree of a Chinese family affected by HCM. Squares represent male family members, circles represent female family members, a slash symbol represents an individual who has died, solid symbols represent affected members, open symbols represent unaffected members, and shaded symbols represent members who are mutation carriers.
Table 1. Clinical characterization of the individuals in the family with TnT gene mutation
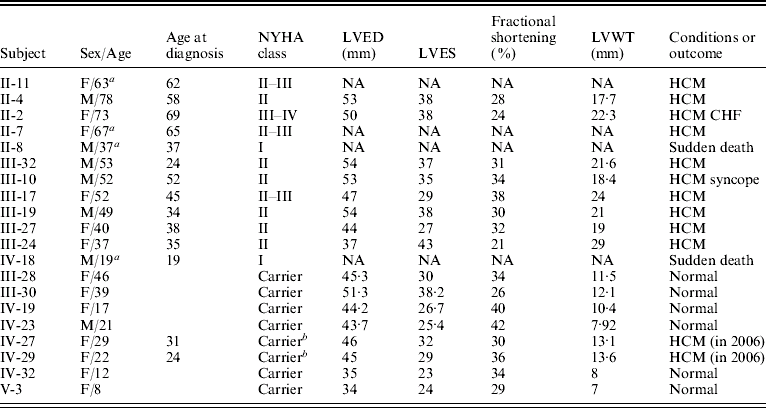
Abbreviations: NYHA, the New York Heart Association; NA, data not obtained; LVED, left ventricular end-diastolic dimension; LVES, left ventricular end-systolic dimension; LVWT, maximal left ventricular wall thickness; CHF, congestive heart failure.
a Patient had died.
b Individuals with the mutation of the TnT gene were healthy subjects in 2004, but were diagnosed as patients with FHCM in 2006 according to the criteria.
Through genome-wide screening, a two-point LOD score of 3·45 (θ=0·15), suggestive of linkage, was initially obtained with the micro-satellite marker, D1S249, and linkage to all other FHCM-causing loci was excluded. A positive LOD score was also obtained with markers flanking D1S249 (data not shown) and this confirmed our finding. Thus, the aetiological gene of this Chinese family affected with HCM was mapped to chromosome 1q32 near the marker D1S249. As the cTnT gene, which is one of the aetiological genes for FHCM, is located in this region, it is reasonable to screen for mutations in this gene. Interestingly, a novel mutation, C→G, at nucleotide 339 in the ninth exon of the cTnT gene (GenBank accession no. NM_001001430), led to a predicted amino acid residue change from Ile to Met at codon 90, and was identified in all patients with HCM (Fig. 2). However, among the 51 healthy individuals, eight individuals without obvious cardiac hypertrophy carried this mutation. It was noticed that the mean age of the eight individuals carrying the Ile-to-Met mutation at codon 90 of the cTnT gene was 26 years (24·25±13·07; range: 8–46 years; Table 1). It is interesting that two of eight individuals carrying this mutation were shown to have a left ventricular wall thickness of >13 mm at the end of the 2-year follow-up by echocardiographic analysis, which is in agreement with criteria of patients with HCM. The other six members were very young, mostly <30 years of age. This sequence variation has not been described in previous studies involving patients with hypertrophic or dilated cardiomyopathies. Furthermore, in an evaluation of 200 unrelated healthy individuals, no subject with this base variation was detected. In addition, the amino acid substituted in human cTnT is strictly conserved across many species, including mice, rats, chickens, quail and zebrafish (Table 2).

Fig. 2. A mutation in the cTnT gene was identified in the patients with FHCM in this pedigree. The mutation, Ile90Met, is caused by a C→G transition at nucleotide 6 of exon 9.
Table 2. Conservation of the TnT residues affected by missense mutations
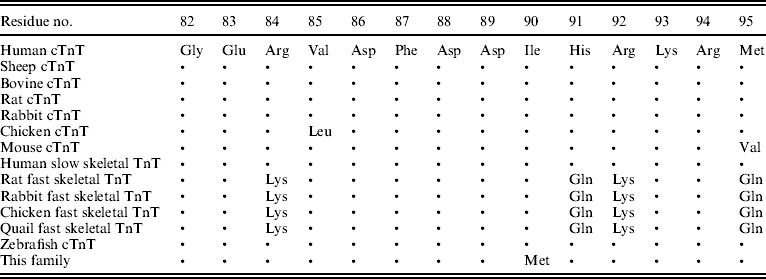
Note: ‘•’ indicates amino acid identity. All protein sequences were obtained from the NCBI Database (http://www.ncbi.nlm.nih.gov/).
4. Discussion
In our study, we demonstrated that a novel missense mutation, Ile90Met, in the cTnT gene may play a critical role in a Chinese pedigree affected by HCM. The sequence variant is believed to be a disease-causing mutation on the basis of several findings. First, through genome-wide screening, a two-point LOD score of 3·45 (θ=0·15) was obtained with a micro-satellite marker, D1S249. The disease-causing gene was mapped to chromosome 1q32 near the marker D1S249. It is well known that the cTnT gene, which is a causative gene for FHCM, was located in this region (Thierfelder et al., Reference Thierfelder, Watkins, MacRae, Lamas, McKenna, Vosberg, Seidman and Seidman1994). Secondly, the nucleotide change identified was present in all definitely affected family members. Although eight carriers had not shown evidence of ventricular hypertrophy initially, 2 years later, two of the carriers were diagnosed as patients with HCM. The other family members may have been too young to exhibit hypertrophy because of the low penetrance of cTnT-causing FHCM. In fact, previous studies have shown that the penetrance of HCM is associated with age. Charron et al. (Reference Charron, Carrier, Dubourg, Tesson, Desnos, Richard, Bonne, Guicheney, Hainque, Bouhour, Mallet, Feingold, Schwartz and Komajda1997) reported that the penetrance of HCM was only 55% in the 10–29 year age group, 75% in the 30–49 year age group, and 95% in the 50-year and older age group. Thirdly, the variant affects a residue that is conserved among all known vertebrate TnT sequences. Fourthly, the Ile90Met mutant at codon 90 of the TnT gene is expected to have a significant impact on the structure and function of this protein, because isoleucine is a non-polar hydrophobic amino acid, whereas methionine is a polar-neutral amino acid. It is located in the Tm-binding domain of human TnT protein and is conserved across many species, including mice, rats, chicken, quail and zebrafish. A small number of the mutations, such as R92L, R92W, R94L, A104V and F110I, in the Tm-binding domain of human TnT protein have been shown to decrease TnT's affinity for α-tropomyosin in previous studies (Palm et al., Reference Palm, Graboski, Hitchcock-DeGregori and Greenfield2001). Thus, the Ile90Met mutant at codon 90 of the TnT gene may be expected to alter the ability of tropomyosin to bind tightly to actin and negatively affect myofibrillar assembly, cooperativity and maximal force and Ca2+-dependence of contraction.
HCM is the most common genetic cardiovascular disease, with reports from many countries (Marian & Roberts, Reference Marian and Roberts2001). Genes involved in this disease all encode components of the contractile apparatus of the cardiac myocyte. Genotype–phenotype analyses revealed some significant differences in prognosis between the patients with FHCM caused by mutations in the different genes or different mutations in the same gene (Konno et al., Reference Konno, Shimizu, Ino, Fujino, Hayashi, Uchiyama, Kaneda, Inoue, Masuda and Mabuchi2005). The distribution of the disease-causing genes has indicated that the β-myosin heavy chain was the most frequent (approximately 35%), followed by myosin-binding protein C (approximately 20%) and cTnT (15–20%). The characteristics of cTnT-causing HCM are mild ventricular hypertrophy (mean maximal left ventricular wall thickness: 11·13±5·14 mm), low penetrance (40% detected by echocardiography) and a high incidence of SCD (mean age at death: 17±9 years) (Moolman et al., Reference Moolman, Corfield, Posen, Ngumbela, Seidman, Brink and Watkins1997). So, it is reasonable that parts of genetically affected individuals are phenotypically healthy, especially youth under 30 years of age. In our study, eight family members bearing Ile90Met had not shown evidence of ventricular hypertrophy initially, but 2 years later, two family members were diagnosed with HCM. This provides further evidence for our conclusion. The other six family members were very young and subject V-1 was only 8 years of age. HCM may not occur at these early ages, but carriers may be expected to develop HCM as they age, although the timing of symptoms and the penetrance of each particular mutation are currently unknown. Therefore, evaluation and follow-up should be carried out by qualified specialists. Physical examinations, electrocardiograms and echocardiograms should be routinely performed. Many risk factors should be avoided or controlled, such as competitive sports, strenuous exercise or hypertension.
TnT is a key regulator of cardiac contraction. Recent results suggest that TnT has a modulatory role as well as a structural role. It has two domains (Fig. 3 A). The N-terminal domain (T1 domain) attaches the troponin complex to tropomyosin and is responsible for cooperative transitions of the thin filament, whereas the C-terminal domain (T2 domain) interacts with troponins I–C (Sehnert et al., Reference Sehnert, Huq, Weinstein, Walker, Fishman and Stainier2002). TnT mutations are clustered in the two domains (Fig. 3 B). In our study, the identified mutation, Ile90Met, was located in the N-terminal domain. Experiments using peptides expressed in Escherichia coli have shown that this portion of the N-terminal tail (residues 70–170) is crucial for TnT binding to tropomyosin, tropomyosin binding to actin and stabilization of the complex with overlapping tropomyosin extremities. Tardiff et al. (Reference Tardiff, Hewett, Palmer, Olsson, Factor, Moore, Robbins and Leinwand1999) showed that isolated cardiac myocytes from R92Q mice have increased basal sarcomeric activation, impaired relaxation and shorter sarcomere lengths. The novel mutation, Ile90Met, is close to R92Q, and as predicted by bioinformatics, the second structure of TnT is α-helical from residue 90. Thus, we suggest that it shares the same mechanism for HCM with its flanking mutations. Furthermore, at the nucleotide level, cytosine is the most commonly occurring nucleotide that is mutated and methylation of this nucleotide has recently been shown to confer instability on the cTnT gene (D'Cruz et al., Reference D'Cruz, Baboonian, Phillimore, Taylor, Elliott, Varnava, Davison, McKenna and Carter2000). In addition, the residue Ile90 is conserved among all known vertebrate TnT sequences. So, we presumed Ile90Met is an FHCM-causing mutation for this Chinese pedigree. Further experiments are needed to explore the mechanism of HCM caused by this mutation.
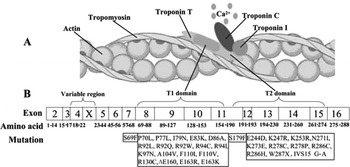
Fig. 3. The model of the thin filament of the cardiac sarcomere and the mutations causing the FHCM have been identified in the TnT gene. (A) Structural relationships of proteins constituting the thin filament of the cardiac sarcomere. (B) Schematic representation of the human cTnT structure with sites of FHCM-causing mutations. T1 domain attaches the troponin complex to tropomyosin and is responsible for cooperative transitions of the thin filament. T2 domain interacts with troponins I–C.
In conclusion, a novel mutation, Ile90Met, in the cTnT gene was identified in a Chinese pedigree affected by HCM. FHCM caused by this mutation showed a relatively moderate left ventricular hypertrophy and a poor prognosis, with two SCDs out of four disease-related deaths. Furthermore, the penetrance of the mutation was not complete (approximately 75%). The development of genotyping in HCM based on a more accurate approach, along with increasing knowledge of the relationship between the genotype and the phenotype, should lead to improved genetic counselling and better clinical management in families with FHCM.
The work was carried out at State Key Laboratory of Medical Genomics. This work was supported in part by grants from the Foundation for the Author of National Excellent Doctoral Dissertation of People's Republic of China (200154; to H.-D. Song), the National Natural Science Foundation of China (30530370, 30771017 and 30470816), and the Commission for Science and Technology of Shanghai.





