



I. Introduction
Software and applications play an ever-increasing role in healthcare and wellness. This is evident with the increased use of Internet of Things (IoT) devices, applications on smartphones for both professionals and consumers and even more sophisticated software used by health professionals.Footnote 1 This presents a dilemma for lawmakers as to when this software, now much more widespread than previously, should be regulated in the same way as other medical equipment or “medical devices” (MD). Software can potentially harm or affect humans in the same way as physical equipment, which means it must be regulated with the same degree of rigour in cases where this is so. However, determining when software is a MD remains a tricky issue.
In the field of MD regulation, the European Union (EU) and the USA are the two biggest players internationally. In this paper, we concentrate on the EU and attempt to identify when software can be considered a MD. There is a range of reasons why it must be clear when software is and is not a MD, with the most urgent being the increased risks that software used as a MD pose.Footnote 2 Insecure software risks the mental and physical health of human beings. Security is a complex process that requires significant safety capital upfront during the design stage, just like conventional MDs. Unlike conventional MDs, software as a MD also requires continuous updates to maintain security, which means a significant upfront as well as continuous capital outlay. Aside from economics, market incentives and a lack of regulation of software combine together to create an environment where security engineering is sadly not of a high standard.Footnote 3
Security failuresFootnote 4 can cause harm when the software is essential for implantable and devices that otherwise affect the physical health of a human (such as pacemakers), and these are also known to have poor defences.Footnote 5 Using secure communication channels, robustness to malware and other harmful interference should be required, but there is no such explicit legal or practical requirement for software in general or MD software specifically, with no certification or authority investigating or inspecting this specific issue at the EU level.Footnote 6 There are also no requirements for ensuring software correctness and that the software is built to a high standard.Footnote 7 Before considering whether the existing legal requirements do take account of these challenges, current research on guidance derived from EU law for software is promising,Footnote 8 in the sense that the guidance does consider security failures,Footnote 9 because it is implied and accepted as a central requirement for functioning when it comes to MDs. If the software that is considered a MD cannot start or it is constantly compromised by adversaries or malfunctions otherwise, it does not fulfil the basic requirements for MDs.Footnote 10
As a recent example, several software-based solutions were proposed that would use mobility data from smartphones and wearables for disease surveillance. The European Commission issued a Recommendation “on a common Union toolbox for the use of technology and data to combat and exit from the COVID-19 crisis, in particular concerning mobile applications and the use of anonymised mobility data”.Footnote 11 This was released after the World Health Organization (WHO) declared COVID-19 to be a pandemic on 11 March 2020.Footnote 12 Many organisations may have developed smartphone-based contact tracing methods without much consideration of the relevant regulatory landscape as a response to the pandemic. Data protection and privacy concerns have been the focus when contact tracing methods were analysed,Footnote 13 but very little regard has been paid so far to MD regulatory frameworks.
Among the many consequences of the virus was the postponement of Regulation 2017/745 – the Medical Device Regulation (MDR),Footnote 14 which is also a primary focus of this paper. It was postponed for a year and came into force on 26 May 2021. Before it was postponed, the European Commission issued the Recommendation on COVID-19 technology and data. In its Preamble 13, it states that the MDR, as well as the Medical Device Directive (MDD),Footnote 15 which was in force at the time, might apply to some of the mobile applications that could be used for the diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease during the pandemic.Footnote 16 This could include self-diagnosis software, and the Commission directly asked for stakeholders to consider whether this type of software falls within the scope of the MDD and MDR.
This paper focuses on this ambiguity implicit in the questions from the European Commission in its Recommendation that is at the heart of EU MD law: namely, the situations in which software falls within the scope of the MDR. We examine shortly how the MDD addressed this, as well as how it is addressed by the MDR. This paper also aims to demonstrate ways to identify software that should be MDs through different means of interpreting EU law in order to prevent circumvention and with the protection of patients in mind. This comes down to a new interpretation of a single term, “intended use”, which we argue needs to be expanded.
The analysis goes through the relevant legislation and concepts, including a framework for understanding intention, a core requirement for MDs. Thereafter, we describe how software as MDs can be identified, and we develop a decision diagram that concretises the identification process in order to provide more clarity and certainty for determining the identification of software as MDs, as well as stymieing attempts to circumvent the regulation.
II. Legal sources, software and its use in the medical industry
Surgical robots, pacemakers, automated insulin dispensers and a large range of other medical equipment that all make use of software in one way or another are at an all-time high. In the EU context, this is not unfamiliar or unusual; however, many manufacturers, caregivers and users may be unaware of the MDR. From late May 2021, all MDs in the EU must comply with it. The use of software is very distinct when it comes to MDs, because of its lack of a physical presence. It may manifest its existence physically, if it is part of a device (an accessory) or if it is necessary for it to function,Footnote 17 and it may also be standalone (ie non-physical) and yet still be a MD.
Increased use of software as MDs, either directly or acting as an accessory, warrants more scrutiny of its structure and usage. Unlike the usual engineering or pharmaceutical perspectives on MDs,Footnote 18 software has security and privacy angles that ought to be considered. Meanwhile, the same considerations overall for MDs must still be taken into consideration. Therefore, interdisciplinary approaches are not novel in this field. Manufacturers and users (medical professionals) increasingly choose to use software in the healthcare sector,Footnote 19 with the software either itself being the MD or for use with a MD (as an accessory), thereby fulfilling the role of medical equipment. Increased use of IoT or robotsFootnote 20 also comes with increased risks in the form of security and privacy breaches. A security breach may include physical harm to the user, which was discussed extensively in an earlier paper.Footnote 21 Likewise, privacy breaches come with their own consequences in the form of theft of personal data, either to sell or to use for more sinister purposes.Footnote 22 Both of these risks are also carried into the MD field from their software origins.
1. Medical Device Directive
MDs are, as of the time of writing, no longer governed by the MDD. From the end of May 2021, the MDD became obsolete, but it continues to be a source of regulatory inspiration because of its similarities to the MDR and its historical significance, so we address it here.
The term “software” is only mentioned twice in the Directive. One of these places is in Article 1(2), where software is included if it is “necessary for proper application intended by the manufacturer to be used for human beings”.Footnote 23 Any software then has to be an “instrument, apparatus, appliance, material or other article”, and it must be intended for certain purposes. These are:
-
(1) Diagnosis, prevention, monitoring, treatment or alleviation of disease;
-
(2) Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap;
-
(3) Investigations, replacement or modification of the anatomy or of a physiological process;
-
(4) Control of conceptions.
These must not achieve their main goals in/on the human body by pharmacological/immunological/metabolic means,Footnote 24 and this is because these devices may be regulated by the in vitro MD regulation (or IVDR) instead.Footnote 25 Software must therefore be treated and evaluated according to the same rules as for every other MD, if covered by the jurisdiction of the MDD. Software can also be considered an accessory to a MD, and in such cases must fulfil the same requirements as the MD (see Article 1(2)(b)). Unlike the Regulation, the MDD was a directive, which entailed a different implementation of the Directive in each EU Member State.Footnote 26 We recognise that some of the criteria are similar to what we see in the new Regulation. However, given the direct effect of the Regulation’s provisions compared to the transposition of the Directive into national legal systems, there may be divergence between past practice under the MDD and new practice under the MDR within a particular Member State’s national jurisdiction.
There is no explicit article on the scope and subject in the MDD, but Article 2 shows that a primary purpose of the Directive is the placement onto the market of MDs, but only if they do not compromise the health of the patients, users or other persons when used for their intended purposes. The rest of the structure resembles that of the MDR, in a shorter and more concise form.Footnote 27 Since the MDR is more comprehensive, we give a more detailed overview of it here.Footnote 28
2. Medical Device Regulation
As an EU regulation is applicable directly through its literal wording, unlike directives which are required to be implemented into national law,Footnote 29 the MDR must be understood as it is. The scope of the Regulation seen in Article 1(1) is to define the rules for MDs in the three senses of “placing on the market, making available on the market, or deploying medical devices and accessories for human use”.Footnote 30 This means that the subjects and the core focus of the Regulation are the devices, and therefore also their manufacturers and anyone else that sells or distributes them. They must as subjects of the Regulation play a substantial part in complying with the rules, self-evidently identifying and loyally fulfilling their duties, which follow from Article 1(1) and Article 10(1).Footnote 31 The jurisdiction of the MDR vis-à-vis manufacturers is potentially worldwide. According to Article 1(1), any manufacturer – seemingly regardless of where they are located – that wants to enter the European Single Market must conform to the MDR. The term “software” is mentioned in the Regulation,Footnote 32 but it is not treated separately, nor are there any specific articles concerning it. The general rules for MDs therefore apply to software. Article 1(1), on the subject matter and scope in the Regulation, does not literally exclude software as being considered a “MD for human use”, and it is not excluded in Article 1(6) either. In the Regulation’s definitions in Article 2(1), software is mentioned as a MD if it used for either or several of the following purposes:Footnote 33
-
(1) Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease;
-
(2) Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or disability;
-
(3) Investigation, replacement or modification of the anatomy or of a physiological or pathological process or state;
-
(4) Providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations.
As with the MDD, software can also be an accessory to a MD (see Article 2(1)). The MDR makes a sharp distinction between software intended for use with or as a MD and software for general purposes (Preamble 19). If not intended to be used with or as a MD, “generic software”, which we define as software for general purposes, can never be an accessory or a MD. If it is specifically intended by the manufacturer to be used as such (but is not generic), it may be considered a MD or an accessory if it fulfils the other requirements. Software that is a MD or is an accessory must also have and display a CE marking (Article 20).
Like with the MDD, before Article 2(1) applies, the software must be intended by the manufacturer to be used for the listed means. This makes the evaluation of whether there exists such an intention a gatekeeper as to whether the MDR even applies in the first place.
In the view of the safety concerns of the public and manufacturers, the character of the MD is to be assessed before it is released onto the market. This is done through classes that designate increasing levels of risk. MDs are divided into class I, IIa, IIb and III, as per Article 51(1). The determinations of these classes are defined in Annex VIII in the MDR. The difference in class can be seen as reflecting the danger a MD poses to those it is used on (be it patients or users), which results in special rules given to the higher classes. For class III, this would include Articles 27, 32, 52, 54, 55, 61, 86 and 105.Footnote 34 Software is mentioned in Section 3.3 of Annex VIII, which dictates that if it drives or influences a device it shall have the same class: this is the main rule.
However, even if the software is independent, it must still be classified with regard to its potential danger to the workflow that can affect the patient. Specific rules follow these two principles. Rule 11 in Annex VIII assumes at first that all software that is also a MD is class I.Footnote 35 The exception to this is software that is used to take decisions on diagnosis or has therapeutic purposes. These are instead considered to be class IIa. The exception to the exception would be if the software can cause serious deterioration of the health of a person, which makes it class IIb, or if it is capable of causing death or irreversible deterioration to the health of a person, in which case it is class III. If the software monitors physiological processes, it is class IIa, but if it can cause immediate harm to the patient during its use, it is to be considered class IIb.
The MD regulatory framework in the EU is enforced by nationally appointed regulators in each Member State (see Article 101). The Medical Device Coordination Group (MDCG), an innovation of the MDR (Article 103), is the new organ that facilitates cooperation between the different regulators, but it is not by itself the central authority. The national authorities can make use of a wide array of enforcement tools, including forceful withdrawal and banning of a MD, as seen in Article 10(14). For the purposes of this paper, the market surveillance activities done by the regulators seen in Article 93 are central as well. This kind of surveillance can include a focus on software distribution platforms, and while it focuses on devices,Footnote 36 this does exclude evaluations of whether software already considered MDs cannot later be excluded based on not fulfilling Article 2(1), especially with a focus on intention. We searched for further information about national authorities’ practices in utilising their regulatory powers under the MDD; however, information about these practices is either not publicly available or does not exist, which makes evaluation of their past regulatory behaviour and prediction of future behaviour difficult. Whether and how the regulators evaluate the intention of the manufacturer ex ante is also unclear and unknown.
National regulators in the EU do not always assess the conformity of the devices initially before they enter the market. This activity is at first left to something called a “notified body” (see Articles 35 and 36). These are usually private companies, which are given the competence to assess MDs.Footnote 37 Software submitted to a notified body may confirm the intention of the manufacturer, as submitting one’s product to such a body is akin to admitting it is a MD directly or implicitly.Footnote 38
3. MEDDEV 2.1/6 and MDCG 2019-11Footnote 39
Issued with the MDD is the “Guidelines on the Qualification and Classification of Stand Alone Software Used in Healthcare within the Regulatory Framework of Medical Devices”, known as MEDDEV 2.1/6 for short. We only highlight points that are important to our discussion, as the guidance also contains several outdated passages.
Issued as guidance before the MDR came into force, “Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR”, called MDCG 2019-11, is the updated guidance on software as MDs. We focus on aspects that are relevant for our analysis from this as well.
Since MEDDEV 2.1/6 was built around the MDD, it did not contain many specific articles from which we can draw inspiration. It does add that generic software should not be considered a MD if it is part of several software modules in one unit.Footnote 40 Every evaluation of such software must be individual (for each type of software), and so the fact that one module is a MD does not also make the entire system one as such. A central limitation of this guidance is that it only applies to standalone softwareFootnote 41 and not software incorporated in MDs such as those seen in surgical robots. However, this does not limit its role for inspiration for interpreting the MDR in the future, together with the new guidance. MEDDEV 2.1/6 assures that the intent of the manufacturer plays a central role, regardless of what the software is called.Footnote 42 This can be interpreted as the situation where the software may be called something in regards to healthcare, but the intent from the manufacturer was purely for the software to act as something informal. The choice of operating system that the software runs on does not affect the evaluation of the software either, and the risk of malfunction of software run in a medical environment does not turn it into a MD per se.Footnote 43
The guidance includes a “decision diagram to assist qualification of software as medical device”.Footnote 44 The MDCG 2019-11 has a slightly updated diagramFootnote 45 that we also use as inspiration. We choose to make use of this decision diagram for our own proposed set of requirements later, but first we need to see how each point of this diagram relates to the rest. The diagram states that if the software does not fulfil the criteria, it is not covered by the MD directives, and we interpret that as meaning that the software is not considered to be a MD. This has been clarified in the new guidance.
Here we identify the aspects of the diagram relevant for this paper’s analysis:
-
(1) The first part relates to whether the software can be defined as software in the guidance. Related to the MDR, there is no stringent definition of this. This is in itself tautological.
-
(2) The second part relates to whether the software is standalone or not. If it is not, it can either be part of a MD or not covered by the MDD.
-
(3) The third part defines that if the software merely acts on data from storage, archives, communication or simple search, it is not a MD.
-
(4) The fourth part postulates that if the actions of the software are not for the benefit of individual patients, it is not a MD. Note that neither the MDD nor the MDR has the benefit of patients among their explicit main purposes.Footnote 46
-
(5) The fifth part says that if the software does not fit a purpose contained in Article 1(2)(a) of the MDD, it is not a MD, and it is if it does. But even if it is not a MD, the software can still be considered an accessory, which would lead to the same requirements as that of a MD.
-
(6) The sixth part says that in order to be an accessory, the software must fulfil Article 1(2)(b), which means that it must be intended specifically to be used with a MD by the manufacturer. Otherwise, it is not a MD or an accessory at all.
As the keen reader would have noticed, while the whole diagram is interesting for inspirational and historical reasons, the fourth part stands out. The MDR does not include as a criterion as to whether a MD benefits individuals; it is not even mentioned literally in the text. It instead considers whether the devices are for human use.
MDCG 2019-11 repeats several of these requirements in its diagram and text, but it is notably different. MDCG 2019-11 does not view software in modules, and instead focuses on whether it fulfils the requirements to be a MD by itself, if it drives or influences a MD, regardless of where it is, and if it is used explicitly for a MD-related purpose by staff/laypersons.Footnote 47 This last point is unique and has no basis in the MDR. The guidance further simplifies the decision diagram from MEDDEV 2.1/6,Footnote 48 and differs with this and the lack of reference to the MDR’s Article 2(1) – instead opting to refer to its own definition of MD software.Footnote 49
4. Case law: C-329/16 SNITEM Footnote 50
Before we create our framework, we need to analyse one central case for the use of this kind of guidance and software as MD in general. A fair number of cases have been decided on by the Court of Justice of the European Union (CJEU) in regards to MDs.Footnote 51 However, most of these do not concern software, but we do have one that has an influence on both the MDD and the MDR. The case, C-329/16 SNITEM, was a preliminary ruling by the CJEU on the correct interpretation of the MDD. The recent nature of the ruling makes the decision of great relevance for the MDR and its interpretation. The case concerned one question with two core points.
First, whether standalone software that gave “medico-social establishment support for determining a drug prescription” could show that it was a MD.Footnote 52
Second, whether a MD has to act in or on the human body to be considered a MD.Footnote 53
The CJEU answered both questions in full. According to the CJEU, the firstFootnote 54 rests on what the manufacturer intended for the software to be used as,Footnote 55 and therefore whether it fulfils one or several purposes in MDD Article 1(2).Footnote 56 If the manufacturer intended for it to be used in this manner, it is a MD. Like today, generic software that is used in a medical setting is also clearly not a MD. Since the software supports the doctor with decision-making in a manner that could affect the patient, it must be considered a MD.
The second questionFootnote 57 was answered with the same legal sources as above. The Court emphasised that there is no difference as to whether the software has contact with a human or not, but rather what the intention of the software is (ie the intention of the manufacturer for it to become a MD).
The case may seem obvious with the event of the MDR, but because the MDD was a directive, it relied on national implementation and interpretation, and while this case came through relatively late, it cemented the importance of the manufacturer’s intention in the partially fragmented state that the MDD was in.Footnote 58
III. The intention of the manufacturer
In this section, we create a framework to measure the manufacturer’s intention for their software to be a MD. This is necessary because of the intrinsic values that define software compared to other types of MD. A scalpel or a syringe is not capable of collecting personal data or independently controlling itself, but many types of software are.
1. Preliminary comments
The manufacturer’s intention, whether stated or otherwise, is central as to whether the MDR applies in the first place, which is seen in the definition of MDs in Article 2(1) and requires the manufacturer to have intended for their product to fulfil one of these enumerated purposes. This was emphasised in C-329/16. In the MDR this is further defined in Article 2(12), which states that the labelling, instructions for use, promotional material or statements made by the manufacturer in clinical evaluation are considered “intended use”.Footnote 59
But how we are supposed to deduce intention, besides from when it is written literally, is not clear from the MDR, nor its preparatory materials, nor past practice or the guidance. Article 2(12) does not go into detail about actual use or how the manufacturers designed the software.Footnote 60
We therefore propose a framework that can aid with this determination and increase clarity and certainty for software manufacturers and ensure consistency for regulators.
The reason we assume that degrees or other abstractions of the intention are needed relates to the special attributes of software. Unlike other medical equipment, the use and the design of software can be deceptive.Footnote 61 This refers to the hidden layers and the ethereal nature of software. Physical appliances (unless they also include software) have no hidden features such as data collection or risks of cybersecurity breaches, but software does, so this framework is made to aid in identifying these as well.
For the MDR to be effective, it must cover all MDs possible, and this framework may help all stakeholders to achieve this. Of course, this framework lists what we consider to be apparent indicative sources for determining the manufacturer’s intention in regards to software as MDs, and therefore it cannot be considered to represent an exhaustive list.
The other reason as to why we must include more considerations for intention is because manufacturers stating that their software is not supposed to be a MD or not mentioning it at all serves as the easiest way of preventing the MDR from applying. Going by the definition in Article 2(12), if manufacturers directly state that their software is not used for a purpose from Article 2(1), the software will not be considered a MD. But the software may very well be used by laypersons or medical professionalsFootnote 62 for treatment, or it may be an app from the Google Play store that would fulfil Article 2(1) had the manufacturer labelled it as a MD. If we purely limit intended use to what is available in public knowledge, as Article 2(12) indicates, we allow any manufacturer, especially of software, to circumvent the MDR, which is both a violation of the Regulation as well as a potential danger for patients, despite the MDR not explicitly focusing on the latter.
It is this grey area that we want to shine a light on and assess.
2. Framework for manufacturer’s intention to qualify software as a medical device
Firstly, we must assume that the MDR did not intend for the manufacturer to be able to circumvent the entire Regulation by merely stating that their software is not a MD. Whether the MDR applies or not should instead be decided by the purposesFootnote 63 laid out in Article 2(1). If the Regulation allowed easy circumvention, it would not be able to fulfil its goals in Article 1(1) and would be rendered positively legally redundant.Footnote 64
Secondly, we must expand the term “intention” to the direct intention seen in publicly available documentation and marketing materials issued by the manufacturer that concern the software. This is the concept seen in Article 2(12) partially combined with even more publicly available sources. But we further argue that “intention” must also include the manufacturer’s indirect intention, which is seen in what the software is capable of, what data it retrieves or measures and what kind of analysis or lack thereof the software is able to do. The indirect intention can therefore be considered as to what the software is capable of and what it does in practice.Footnote 65
Indirect intention must be viewed as the actual capabilities of the software,Footnote 66 regardless of what is stated in publicly available sources. To include all possible angles for this evaluation, distinctions between how the data used for usage are gathered and what the software does with the data are necessary. We include a third category to catch software in grey areas: software that acts and functions as a MD already.Footnote 67 However, this is not equivalent to a blanket statement that could include, for example, all smartphone apps just because some existing MDs that are software can run on this specific hardware. Instead, this refers to software that is capable of the same as existing MDs or that users can use directly as substitutes for existing MDs.Footnote 68
We consider that in the case of conflict between the two types of intention, if either indicates the intention of the manufacturer for the software to be a MD, the affirming intention will prevail. It may still after that not be a MD because it does not fulfil any of the purposes in Article 2(1), even if the manufacturer intended for it to do so.
3. The framework
Firstly, we propose the following as sources for discerning direct intention:
-
(1) Information from marketing materials. If the manufacturer states that the software is a MD or claims it fulfils one of the purposes in Article 2(1), direct intention can be established.
-
(2) Information from internal documentation. If the manufacturer states that the software is supposed to fulfil one of the purposes in Article 2(1) in its internal documentation to which the EU Member State national regulators or other public authorities have access due to Article 10 or other legal provisions (eg in national public law), direct intention can be established.
-
(3) Informal information sources. If manufacturers or their representatives have said, expressed or if it is stated in search systems through such media as tags that the software fulfils one of the purposes in Article 2(1), direct intention can also be established.
Secondly, we propose the following as sources for discerning indirect intention:
-
(1) Data-gathering practices. If the software gathers or measures data that are relevant for fulfilling the purposes in Article 2(1), an indirect intention can be established. This can be biometrical data about, as well as health records of, a natural person.
-
(2) Data analysis. If the software, as part of its purpose, requires personal data to be analysed to reach results that resemble or fulfil the purposes in Article 2(1), an indirect intention can be established.
-
(3) Software specifications. If the software is designed and made to function as a MD, with the aim of either substituting or replacing existing MDs without being one itself, an indirect intention can be established.
An illustration of the framework is shown in Figure 1.
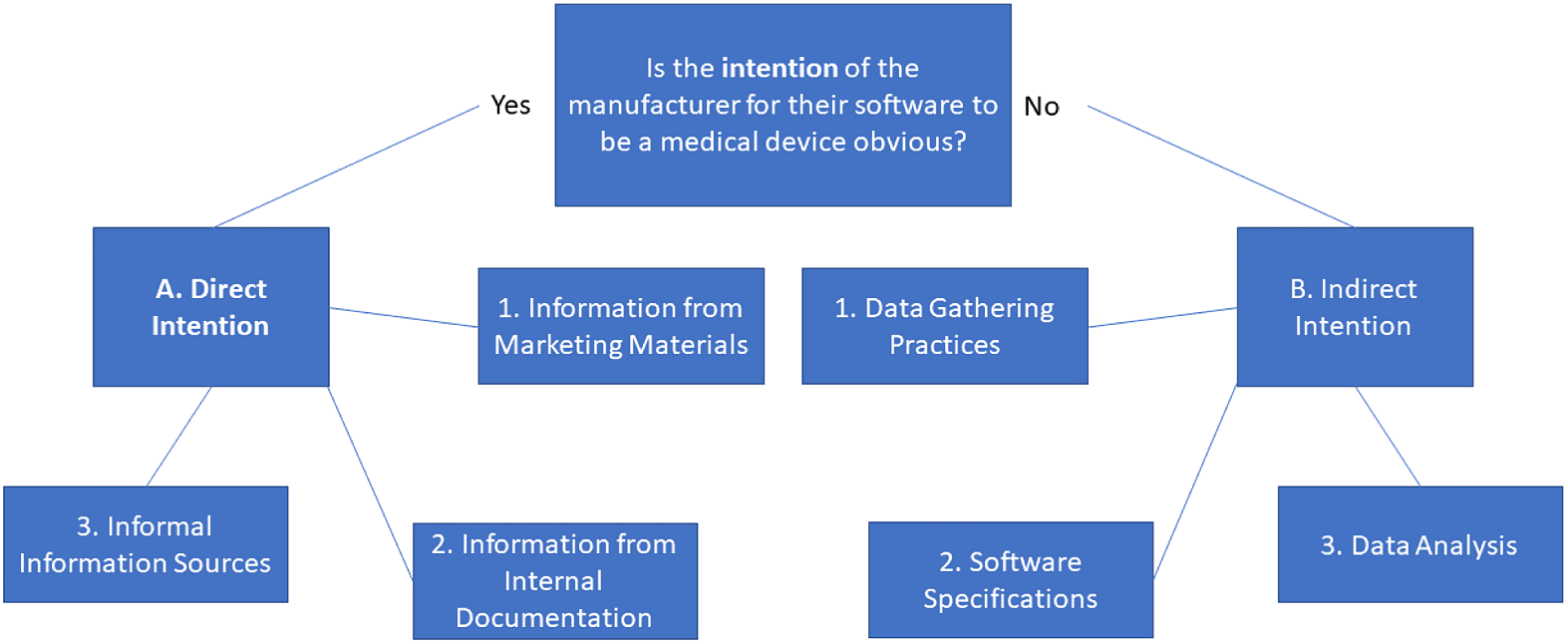
Figure 1: Diagram of the framework of intention.
IV. Requirements for software to be a medical device
In this section, we propose a set of more direct requirements for when software is considered a MD or not in the regime of the MDR. We make the requirements fit the MDR, because it is now the active legislation, while the MDD is now inactive.
1. Overarching concepts
We first make three necessary distinctions. These are: whether the intention of the manufacturer is clear; whether the software is generic; and whether the software is an accessory.
-
(1) If the software is not intended for one or more of the medical purposes in Article 2(1), it cannot be considered a MD. This is subjective, and we showed in our framework that the intention can be direct or indirect. Because of the wording in the MDR, resolving whether there is intention or not must be done before anything else.
-
(2) If the software is generic and not modified by the manufacturer to be used standalone or as in a module/system, it is not a MD.Footnote 69
-
(3) If the software is intended by the manufacturer to be an accessory to a MD, either directly or to assist it, the software cannot by itself be considered a MD. It is considered an accessory instead.
We know from the CJEU’s decision in C-329/16 that there exists a fourth requirement:
-
(4) If the software is not used for or on human beings, it cannot be a MD.
2. Considerations for the requirements
The fourth point of the decision diagram used for standalone software in MEDDEV 2.1/6 distinguishes between software that stores, archives, communicates or searches in data and software that does other actions. There is no basis for this in the Regulation, and the points above support the idea, since purely storing, archiving, communicating or searching does not fulfil any of the purposes, even if the manufacturer may intend it to do so.
As these are points of interpretation, certain considerations have to be taken account of for each. All points refer to software that specifically is intended to be used for such purposes; this rules out software that merely facilitates and is specifically not intended for these purposes. However, it also catches software that merely monitors disease and disability.Footnote 70
Alleviation of disability does not exist, but compensation is possible – which means that software that, for example, is used to control an artificial limb is either a MD or an accessory to a MD. It includes replacement of limbs or organs as part of “replacement or modification”, even if the software merely calculates movements or facilitates pumps. This part is extremely wide due to it covering all physiological and pathological processes or states.
When “used” is written, we can refer back to C-329/16, which, in Paragraphs 30 and 32, assures us that the software does not need to physically be “used” on the human body. This again broadens the range of software that may then be considered as a MD. But as broad as it sounds, we still want to include a criterion that covers and forces the software to be used on/for humans, even if there does not exist a physical requirement as such, which is in line with C-329/16.
The framework of intention plays a major role (see Article 2(1) in the MDR) as to whether we can even start using the requirements. While the MDR does not explicitly say that the regulators are to search and survey the software market to discover whether software could be a MD or not, they are not prevented in doing so. Their search could therefore include all software clearly marketed as such and all software that clearly acts as a MD.Footnote 71 This kind of search could be encompassed by the obligations of regulators.Footnote 72 We illustrate the requirements discussed above as a decision tree, which is shown in Figure 2.
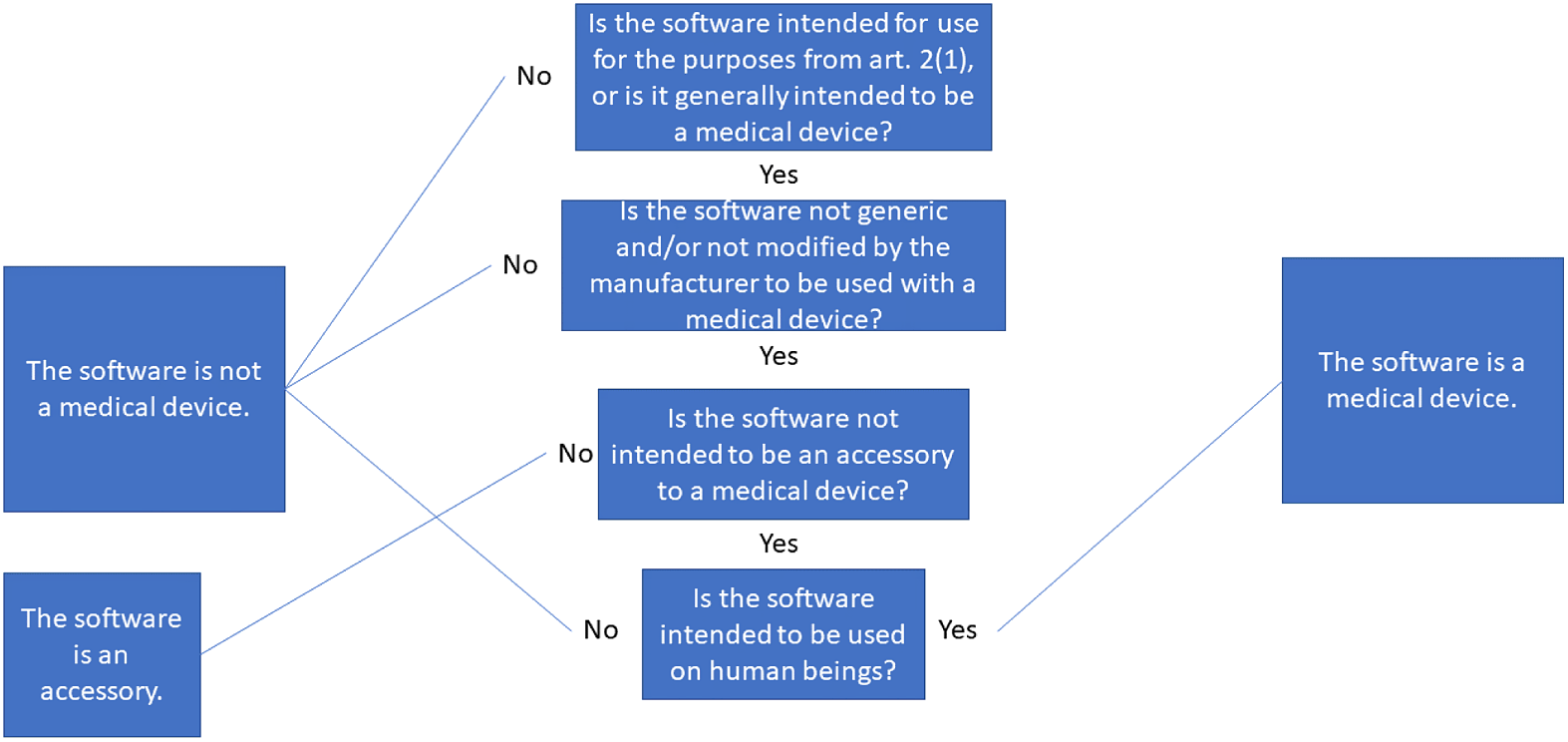
Figure 2: Illustration of the requirements for software to be considered a medical device.
VI. Concluding remarks
Software is here to stay in MD regulation, but the event horizon that is apps and the IoT is arriving fast, and in fact is already present. With our suggestion to expand the idea and terminology so that intention includes what software does in practice, we hope that the MDR can cover and increase the quality of software as MDs in the future, both in the EU and in the world as a whole.
VII. Next steps
This article is limited in its focus and further research on software and MDs is necessary. This could include the analysis of specific genres of software and how they would be treated by the MDR. A specific example relates to applications that focus on physiological diseases or disabilities or act purely as accessories, which could be analysed to show how the requirements framework is applied in practice. Further research is also needed on the actual practices of EU MD regulatory bodies through, for example, expert interviews. Furthermore, policy research that addresses the enforcement issues we have identified would be useful, as would research that could show a different interpretative angle regarding the MDR in general. And finally, empirical research in needed on which apps on smartphones could be considered a MD if one uses indirect intention to determine it.



 Open access
Open access