


Measles is a highly infectious virus with the potential to cause pneumonia and encephalitis, which may lead to death. As such, measles is a leading cause of vaccine-preventable childhood mortality. The United Nations (UN) Millennium Development Goals (MDG 4) target a two thirds overall reduction of child deaths by 2015 compared to the 1990 level [Reference Duclos1]. At the 63rd World Health Assembly, delegates targeted a 95% reduction in measles mortality by 2015 from 2000 levels, and a move towards its eventual eradication [2]. Projections suggest that the annual number of measles deaths could rebound resulting in about 500 000 deaths in 2013 [2].
The World Health Organization (WHO) and UNICEF have identified 47 priority countries with the highest burden of measles for an accelerated strategy for measles mortality reduction, to include high vaccine coverage and measles surveillance. The WHO Measles and Rubella Laboratory Network [Lab-Net (http://workspace.who.int/sites/genotype)] has been established to monitor progress towards mortality reduction and elimination of measles [Reference Ji3]. In 2008, China was third (behind India and Nigeria) in the number of children who missed receiving their first dose of measles vaccine through routine immunization services [Reference Zhang4]. Therefore, China remains a focus country for sustaining measles control which in addition to increased vaccine coverage, is also dependent on active surveillance reporting.
In the 2005 measles epidemic in China, Jiangsu had the third highest number of measles cases, with the highest number wild-type measles viruses isolated. It was observed that the resurgence of measles was reported to be associated with continued circulation of genotype H1 viruses in China, 2005 [Reference Ji3]. Nantong is a prefecture-level modern port city on the eastern coast of Jiangsu province that suffered a recent outbreak of measles cases. Here we report sequence and phylogenetic analyses of wild-type measles nucleoprotein (N) genes in order to better understand the molecular epidemiology of the outbreak in this region of China.
Throat swab and serum samples were collected from suspected cases from January to December 2010. All suspected patients developed typical clinical symptoms: high fever, cough, conjunctivitis, Koplik's spots on the buccal mucosa, and a rash initially of the face, torso, upper neck and back, spreading eventually to the hands and feet. Venous blood samples were collected to obtain serum for detection of anti-measles IgM antibodies by a commercial ELISA kit (Zhuhai Haitai Co., China). Throat swabs from anti-measles IgM-positive patients were inoculated onto cells for virus isolation.
Measles virus was isolated using the Vero/SLAM cell line. Vero/SLAM cells were grown in DMEM supplemented with 10% heat-inactivated FBS (Invitrogen Corp., USA), 0·15% sodium carbonate and 0·38 mg/ml Geneticin® (Invitrogen Corp.). At 90% confluence, cells were infected with clarified virus transport medium and incubated at 37°C for 2 h then washed with PBS. Cells were incubated with media for a further 7 days and the media harvested and stored immediately at −70°C. Attached cells were harvested with a cell scraper, centrifuged at 3000 g for 10 min, and the supernatant collected and stored at −70°C.
Viral RNA was extracted from infected cell lysates using the QIAamp® Viral RNA kit (Qiagen Co., Germany) according the manufacturer's instructions. The cDNA corresponding to the entire coding region for the carboxyl terminus of N genes were synthesized using an Invitrogen kit (Invitrogen Corp.) with random hexamer primers. Primers MV63 (5′-CCT CGG CCT CTC GCA CCT AGT-3′) and MV60 (5′-GCT ATG CCA TGG GAG TAG GAG TGG-3′) were used to amplify a 676-bp fragment of the nucleoprotein (N) gene including the 450-bp fragment recommended for genotyping. PCR reactions were performed using 2–5 ng cDNA, 0·2 pmol of primers and 2 U of AmpliTaq gold DNA polymerase, 200 μm of each dNTP and 1·5 mm MgCl2. PCR was performed on the Agilent Stratagene Gradient Cycler (Agilent Technologies Inc., USA) with the following PCR cycle parameters: 2 min at 95°C for Hotstart Taq DNA polymerase activation, followed by 40 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 50°C and elongating for 1 min at 72°C. Finally an additional elongation step was performed at 72°C for 10 min.
The PCR products were separated and purified using a commercially available kit and sequence analyses performed using an ABI PRISM® 3130 sequence detection system (Applied Biosystems, USA) with primers MV60. The sequences of all samples were analysed using BioEdit and MEGA3.1 software (Arizona State University, Tempe, AZ, USA). Evolutionary distances were estimated using Kimura's two-parameter method and phylogenetic trees were constructed using the neighbour-joining (NJ) method. The robustness of the groupings was assessed using bootstrapping (1000 replicates) and the tree was visualized using the MEGA program.
There were 205 suspected cases in Nantong city in 2010. Venous blood samples were collected from all of these cases to obtain serum for detection of the anti-measles IgM antibody with ELISA. The incidence of measles in 2010 in the greater Nantong city region was 0·45/100 000 with a total of 34 confirmed cases. The average patient's age was 25 years with nine patients (26·47%) aged <4 years and 23 (67·65%) aged >15 years. Almost one-quarter of patients (8/34, 23·53%) had received measles vaccination (either measles vaccine or the measles-mumps-rubella combination vaccine).
Six wild strains were isolated from the swabs collected from all confirmed cases using the Vero/SLAM cell line in 2010 in the greater Nantong city region. PCR products of all isolates in the COOH-terminus of the nucleoprotein gene were sequenced and analysed. All of the measles strains isolated in this study were clustered within genotype H1. The strains characterized showed the highest degree of identity (99%) with the strain Mvi/Hunan.CHN/, the reference strain of H1 published by WHO.
The results of the phylogenetic analysis of COOH-terminal coding of the nucleoprotein gene together with WHO reference strains are shown in Fig. 1. Five nucleotide sequences obtained were submitted to Genbank. The acquired accession numbers were HQ424900–HQ424904. All genetic changes in the Nantong isolates evaluated in this study were base substitutions. We compared the nucleoprotein gene of all isolated measles virus strains with the reference H1 strain published by WHO and found no deletions, insertions or frame-shift mutations. The homology of the nucleotide sequences of the six isolates was 96·46%.
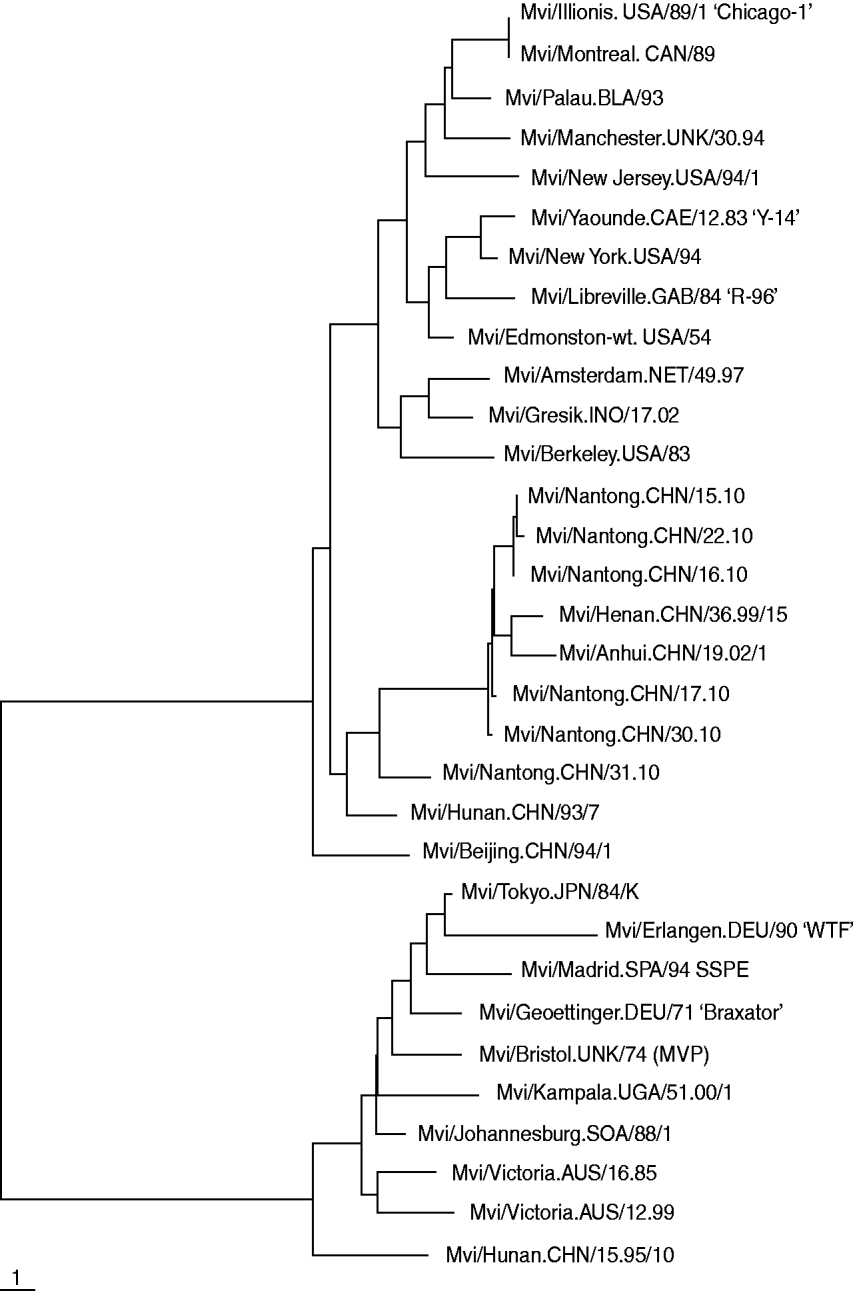
Fig. 1. Phylogenetic relationships based on a 450-bp region between the measles wild strain isolated from Nantong City during 2010 and the WHO reference sequence.
Phylogenetic analysis of the nucleoprotein gene of the isolated wild measles viruses from Nantong city together with the strains from other provinces in China and other Asian countries is shown in Figure 2. The results indicate that the measles viruses isolated in Nantong in 2010 have a high degree of homology with strains detected in other provinces. Compared to strains isolated from other provinces in China in 2007, there were no significant gene mutations, but there was a certain genetic distance. This means that there were different transmission chains and that the same strain was circulating during these years.
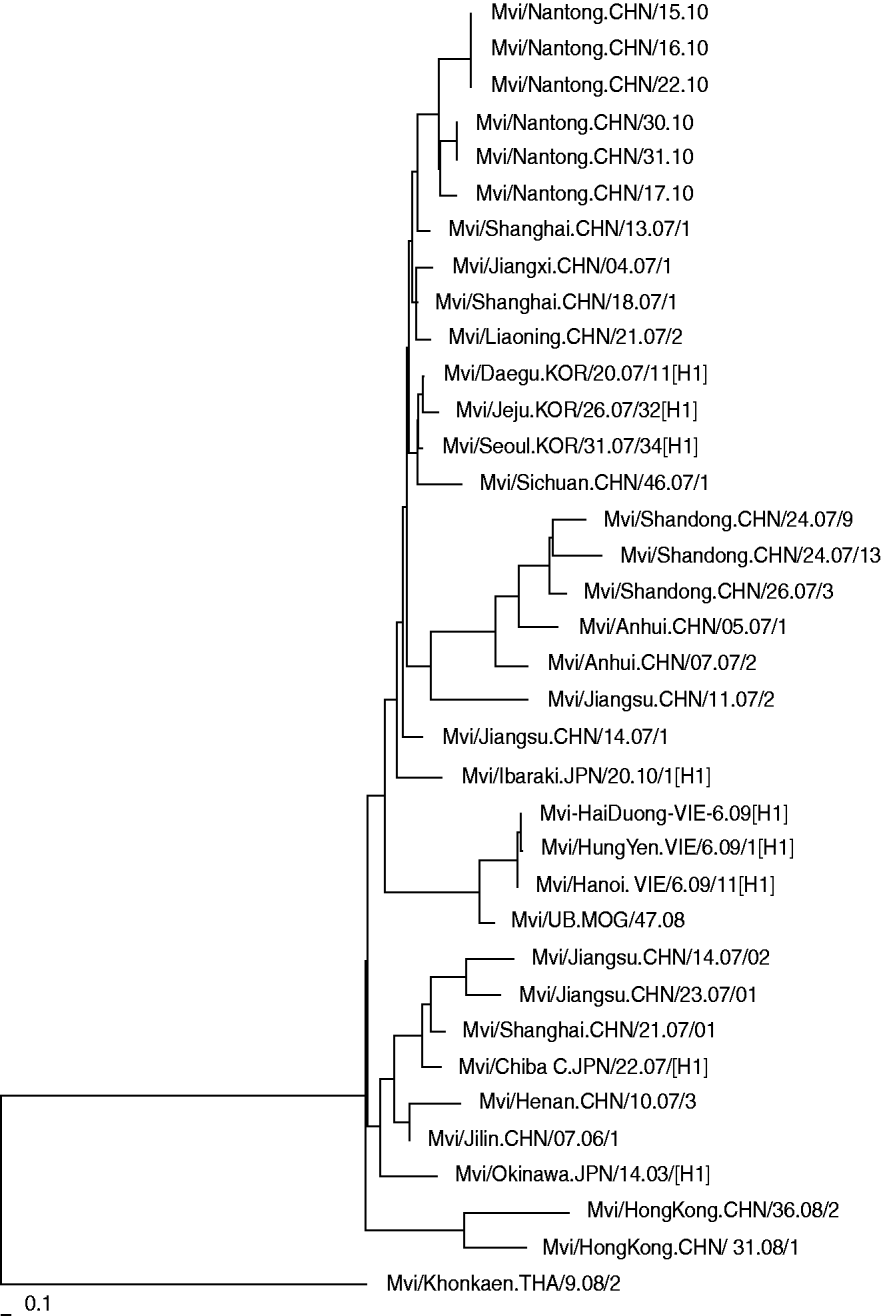
Fig. 2. Phylogenetic relationship between the Nantong isolated measles strains and circulating strains in China and Asian countries.
Viral surveillance during all phases of measles control has been recommended by the WHO [5]. Baseline virological surveillance allows development of accelerated control activities and documentation of changes in viral genotypes can provide evidence of an interruption of endemic transmission of measles. Molecular characterization is therefore an important component of measles surveillance because it enhances the ability to identify the source and trace endemic transmission pathways of the virus. The measles genotypes detected in the Western Pacific Region between 2007 and 2009 were H1, D4, D9 and d11 with H1 associated with endemic transmission and the remaining genotypes associated with importation [Reference Rota6]. In 2006, genotype H1 was widely distributed throughout China with about 100 000 measles cases detected and 356 viruses isolated from September 2006 to August 2007 throughout the country [Reference Ji7]. Multiple transmission pathways were detected, but genetic diversity appeared to decrease compared to the previous 12-month period. The measles cases were all sporadic without any outbreaks in 2010, and the incidence of cases in recent years is declining. The measles viruses were all isolated from the Han population, which is the majority population in China. Nantong is the coastal city closest to Shanghai, the biggest city in China, and therefore has a large floating population. Consequently, it is difficult to identify the origin of the disease. The results here indicated that all of the viruses isolated in this study clustered within genotype H1. These results confirm the endemically circulating MV strain in the coastal region of Jiangsu with low importation rates. This is because the genetic heterogeneity of the Chinese measles viruses is not due to increased mutation rates but rather to the presence of co-circulating virus lineages [Reference Zhang8]. Aggressive measles control strategies are having an impact on genetic diversity and transmission patterns of the measles virus in a coastal region of Jiangsu province, China.
ACKNOWLEDGEMENTS
This study was supported by the financial grants from Jiangsu Prevention Medicine Foundation (YZ201014).
DECLARATION OF INTEREST
None.


