


Introduction
In the famous parable that originates in India of the blind men and the elephant, six blind men lived in a village and experienced the world by hearing stories about it. They learned about many things but become particularly fascinated by the elephant which (in stories they are told) tramples forests, carries huge burdens, and makes a loud trumpet call. But they also hear that the Rajah’s daughter would ride the elephant when she travels around her father’s kingdom. How could this thing be dangerous, if the Rajah lets his daughter ride it, and also be so loud? They would argue about what it was – a powerful giant, a large cow, a graceful ride for a princess, and so on. The villagers grew tired of the argument and arranged to have the blind men examine a real elephant. One touched its side and decided it was smooth and powerful like a wall, as he had believed. Another touched the tusk and announced it was like a sharp spear, as he had believed. Another touched the leg and decided it was nothing more than a large cow, as he had believed. Another touched its trunk and concluded it was a very large snake, as he had believed. And so on. They bickered until the Rajah overheard them. He chastised them for being so certain that they knew what an elephant was – they had each only felt a part of the elephant. Only by putting together all of the pieces could they understand what an elephant was.
The chapters in this part are far from the blind men; they are quite illuminating, indeed, and are very self-aware as to what each of their parts are doing and not doing. But if the elephant represents the legal issues with the way at-home diagnostic devices are actually put on the market, it remains true that each of the first three chapters in this part focus on one piece of the elephant, while the collective helps us better understand it.
David A. Simon and Aaron S. Kesselheim’s “Physician and Device Manufacturer Tort Liability for Remote Patient Monitoring Devices” focuses on the ex post regulation of the US tort system. Focusing in particular on remote patient monitoring (RPM) devices, they examine how current US tort law applies to different players in the RPM device ecosystem: The manufacturers of the devices, physicians who prescribe them, patients who use them, and patients’ caregivers. They also examine the way in which the various regulatory pathways to get an RPM Device to market – premarket notification approval (PMA), the 510(k) and the de novo pathway potentially – preempt certain kinds of tort claims in this space (manufacture, design, marketing) and more specific claim types, such as fraud on the FDA.
In their chapter, “Post-Market Surveillance of Software Medical Devices: Evidence from Regulatory Data,” Alexander O. Everhart and Ariel D. Stern shift to a different ex post form of regulation – post-market surveillance of the subset of RPM and other software-driven products that meet the definition of a medical device in the United States and, therefore, are subject to regulation by the US Food and Drug Administration (FDA). While Simon and Kesselheim’s primary way of touching the elephant is through case law analysis, Everhart and Stern offer an analysis of a large dataset they create. They identify all 510(k)-track and PMA-track medical devices – that is, moderate and high-risk devices – cleared or approved by the FDA from 2008–2018 in five common regulatory medical specialties that are most likely to include RPM devices. In that dataset they identify all the recalls and adverse events associated with these devices that occurred between 2008 and 2020. They find, among other things, that “software-driven medical devices” had higher adverse event and recall probabilities compared to devices without software components. They also argue that for us to truly understand this elephant better, we need a systematic collection of unbiased data describing the post-market performance of both medical devices and digital diagnostics.
Sara Gerke’s chapter, “Labeling of Direct-to-Consumer Medical Artificial Intelligence Applications for ‘Self-Diagnosis,’” shifts from ex post to ex ante regulatory mechanisms, with a focus on direct-to-consumer medical self-diagnosing artificial intelligence (AI) apps. She begins by showing that under the Federal Food, Drug, and Cosmetic Act (FDCA), Congress has given the FDA the power only to regulate software functions that are classified as medical devices under the FDCA. She then examines the guidance promulgated by the FDA for mobile medical apps and “software as a medical device” (SaMD), defined as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” Apple’s electrocardiogram (ECG) and irregular rhythms notification feature apps are good examples of SaMDs. Gerke then takes readers through the exceptions created by Congress through the 21st Century Cures Act to the medical device definition for certain software functions and explains the test the FDA has settled on for determining when it will apply regulatory oversight versus enforcement discretion. The chapter then shifts from the descriptive to the prescriptive in examining the labeling of direct-to-consumer medical self-diagnosing apps as information-only versus diagnostic, and the significant discrepancy between the user’s perception of the intended use of the apps and the intended use put forward by the manufacturer. She argues for labeling standards for AI-based medical devices, including direct-to-consumer medical self-diagnosing AI apps that, among other things, would effectively inform consumers about the type of AI used (e.g., a black box, an adaptive algorithm, etc.), the various risks of bias, the risks of false positive and negative results, and when to seek medical help.
While these three chapters are up-close perspectives on pieces of the legal elephant described through various methods – doctrinal, empirical, regulatory – Zhang Yi and Wang Chenguang’s chapter allows us to view the elephant from afar, by a comparison to how the same issues are handled in a very different legal system: China. In “‘Internet Plus Health Care’ as an Impetus for China’s Health System Reform,” the authors introduce non-Chinese readers to the regulatory category of “internet plus health care” (IPHC), the way that China regards “the use of digital technologies in support of the delivery of health care and health-related services, such as internet-based diagnosis, treatment, and medicine, and internet hospitals.” While these technologies had some support in China even earlier, the 2009 round of health reform really brought them to the fore with increasing initiatives until 2019. The COVID-19 pandemic supercharged interests in these technologies in China, much as it did with telemedicine in the USA, leading the National Health Commission to publish its Regulatory Rules on Internet-based Diagnosis and Treatment in March 2022, among other forms of regulation. The authors then identify some of the remaining challenges – the way the regulations limit IPHC to “follow-up” diagnoses for “common diseases” and “chronic diseases,” the affordability and insurance coverage of IPHC, and the difficulty of translating “physician multi-site practicing” to the online world in a way that is high-quality and accessible.
When read against the first three chapters in this part, the most striking takeaway from this fourth chapter is just how much China treats digital home diagnostics as the regulation of health care as opposed to the regulation of devices. The USA has centralized a lot of the ex ante and ex post regulation to the FDA and to the tort law around the products that is applicable to medical devices in general. In part because of the frequent shibboleth that the FDA does not regulate the practice of medicine, the agency does not directly regulate the integration of digital home diagnostics into health care let alone questions of affordability and insurance coverage. By contrast, it would seem that China views the devices as a means to health care delivery and, thus, leads with that. What would it mean if the US regulatory system started with health care system integration and incorporated that into its regulatory review?
I The Landscape of Remote and Diagnostic Devices
New technologies allow patients to use, wear, or even have implanted remote patient monitoring (RPM) devices that collect data, which can be sent directly to physicians.Footnote 3 These data can be used to identify disease-related events that require medical intervention. RPM includes diagnostics performed by patients at home, without direct physician involvement, that had traditionally been performed in a clinical setting (such as a mobile sleep study), as well as services that combine routine monitoring and diagnosis (such as a heart rate monitor). For example, pacemakers that used to primarily support a patient’s cardiac rhythm can now be used to transmit information to a cardiologist, potentially detecting arrhythmias that may lead to medical treatment at a presymptomatic stage.Footnote 4 Wearable glucose monitors, like Abbott’s FreeStyle Libre 2, and seizure detection devices, like Empatica’s Embrace2, can alert patients or caregivers to low glucose levels and seizure activity that require attention.Footnote 5
With the increasing prevalence of RPM devices, questions remain about the liability protections for patients who use them. State laws, in particular tort law, provide some potential safeguards by enabling patients to sue device manufacturers and physicians for causing them harm. While a variety of state and federal laws impose obligations on manufacturers,Footnote 6 tort law is a major tool to hold these actors accountable for injuries they cause to patients.Footnote 7
The stakes are high. A cardiac monitor or a seizure detection device, like Embrace2, that malfunctions could result in brain damage or death, opening the manufacturer to large jury verdicts, particularly for widely used products. Physicians who improperly use or rely on RPM devices to notify them of such activity and fail to monitor patients could also face substantial damage claims.
Despite the significance of potential injury for patients and liability for manufacturers and physicians, it is not clear how these claims should be evaluated or resolved. To clarify when liability might arise, this chapter first explains how tort liability applies to manufacturers of RPM devices, physicians who prescribe them, and patients who use them (and their caregivers). It then proceeds to analyze how variation in device market entry, patient access, and use – through federal regulatory protections, physician prescriptions for devices, and patient and caregiver uses – can affect the viability of tort claims.
II Liability for Device Manufacturers and Physicians
Tort law contains two primary standards of liability typically applicable to devices like RPM devices (Table 8.1).Footnote 8 The first is negligence, which requires one to act with “reasonable care” when undertaking an activity. For a plaintiff to succeed in a lawsuit based on negligence, the plaintiff must prove that another failed to act with reasonable care, and that such failure caused harm to the plaintiff. The second is strict liability, which does not require such a showing; in theory, there is “no fault” because tort law imposes liability on the person who caused the injury regardless of whether that person acted with reasonable care. Both negligence and strict liability can apply to RPM manufacturers. Typically, only negligence applies to physicians.
Table 8.1 Schematic of tort liability for manufacturers, physicians, and caregivers

This table depicts the potential tort causes of actions against physicians, manufacturers, and caregivers arising from RPM devices. Misrepresentation/fraud claims are depicted in dotted lines to indicate potential causes of action that are not discussed in this chapter.
A Manufacturer Liability for Product Defects
i Negligence
Manufacturers have a duty to use reasonable care in manufacturing, designing, and marketing a product.Footnote 9 They are, therefore, liable for injuries caused to users by failing to reasonably warn of product risks or failing to use reasonable care in designing or manufacturing the product. The standard for negligence claims primarily focuses on the reasonableness of the manufacturer’s behavior. Although evidence of industry custom is admissible in determining the relevant standard of care, industry custom does not determine the relevant standard of care.Footnote 10 That is a determination left to the fact-finder, and if it is a jury, with assistance from the judge.
ii Strict Liability
Manufacturers can also be liable under the theory of strict liability for the same three types of product defects (manufacturing, design, marketing) as they can be liable for in negligence. Unlike negligence, however, strict liability does not require the injured party to prove any negligent conduct by the manufacturer – only that the product defect existed when it left the manufacturer’s hands.Footnote 11 Manufacturing defect claims allege that a defect arose in the production of the product that differed from the manufacturer’s design, and that this defect caused harm to the plaintiff.Footnote 12 Design defect claims allege that, even if manufactured properly, the manufacturer’s design was particularly unsafe and, therefore, defective, and that the defect caused injury to the plaintiff.Footnote 13 Finally, marketing defect claims – also called “failure to warn” or “inadequate warning” claims – allege that the manufacturer failed to provide to the patient with sufficient warnings about the risks of using the product.
iii Scope of Strict Liability Claims
Whether and how negligence or strict liability theories apply can depend on the type of defect alleged, the jurisdiction in which the lawsuit is filed, and the type of product at issue. The type of defect alleged can affect what the plaintiff must prove – with requirements occupying three places along a spectrum. At one end of the spectrum are manufacturing defect claims, for which the only questions are whether the product was manufactured according to the manufacturer’s design and specifications and, if not, whether that defect caused the plaintiff’s injury.Footnote 14 For example, liability under this theory would arise if a patient was injured by a pacemaker that malfunctioned because, during manufacturing, the manufacturer failed to install a computer chip required to process heart rhythms.
At the other end of the spectrum are failure to warn claims, for which the standards for strict liability and negligence are identical – the only question is whether the manufacturer reasonably warned the consumer of the product risks.Footnote 15 For example, a manufacturer of vaginal mesh may be liable on this theory for failing to warn that mesh removal may be required if the product fails.Footnote 16
Somewhere in the middle are design defect claims. Here, the plaintiff must show either that “the product failed to perform as safely as an ordinary consumer would expect when used in an intended or reasonably foreseeable manner” or that “the product’s design proximately caused his injury and the defendant fails to establish, in light of the relevant factors, that, on balance, the benefits of the challenged design outweigh the risk of danger inherent in such design.”Footnote 17 In negligence, courts tend to ask how to balance the device’s risk of harm against its utility, while in strict liability, they tend to emphasize the existence and monetary costs of using an alternative safer design.Footnote 18 For example, the manufacturer of an air conditioning compressor was found liable for injuries caused by an explosion it could have prevented by simply and costlessly relocating a safety groove from the inside to the outside of the compressor’s insulating glass.Footnote 19 In some cases, medical devices like hip implants may be subject to a similar analysis when the device fails.Footnote 20
Jurisdictions may differ, however, on whether strict liability applies. In some jurisdictions, a design defect claim for devices that are “incapable of being made safe for their intended and ordinary use”Footnote 21 will immunize a manufacturer from design defect claims if the manufacturer properly manufactures and warns consumers about the product’s risks.Footnote 22 In such cases, adequate warnings immunize manufacturers from strict liability design defect claims.
All this suggests that the type of device – whether it is “incapable of being made safe” – can also influence whether strict liability applies. Some courts have found that prescription and implantable medical devices count.Footnote 23 Others disagree or think that the question must be resolved on a case-by-case basis by weighing the risk-utility tradeoff presented by the device,Footnote 24, Footnote 25 sometimes casting the issue as one the defendant manufacturer must raise and prove as an affirmative defense.Footnote 26 Finally, there remains something of an open question about whether software itself can be a “product” subject to strict liability.Footnote 27
Device type and jurisdictional issues can also interact to affect potential tort claims. So, even if immunity from strict liability applies, it may apply only to design defect claims (leaving strict liability claims for manufacturing and marketing defects),Footnote 28 or it may bar all strict liability claims.Footnote 29 In some jurisdictions, however, immunity from strict liability claims does not apply to negligence claims.Footnote 30
iv The Learned Intermediary Doctrine
Claim type and use, including the process by which a consumer use occurs, can also affect liability by shifting obligations from one party to another. Marketing defect claims, for instance, require the plaintiff to prove that a product was unreasonably dangerous because it lacked adequate warnings or instructions.Footnote 31 This duty ordinarily requires manufacturers to warn consumers directly. But when a physician prescribes the product, the “learned intermediary doctrine” requires a manufacturer to adequately warn only the prescribing physician subject to three limited exceptions.Footnote 32, Footnote 33
Because warning the physician may require different disclosures than warning a consumer, the learned intermediary doctrine can alter the manufacturer’s explanation of device risks. This can also affect other claims. For example, a manufacturer that successfully defends a failure to warn claim may also be able to defeat liability for a design defect claim, since immunity from some design defect claims requires adequate warnings. At the same time, however, the learned intermediary doctrine will not affect manufacturing defect claims because they do not turn on whether the manufacturer gave proper warnings.
B Physician Liability for Lack of Informed Consent and Negligence
The learned intermediary doctrine is also related to the doctrine of “informed consent,” which imposes on physicians a duty to obtain, prior to treatment, patient consent by informing them of the material risks associated with the treatment. In some jurisdictions, the sufficiency of informed consent is based on whether “the physician’s failure to inform fell below the medical community’s standard of care.”Footnote 34 In others, the question of sufficiency is based on a record of the disclosure of facts that would influence the patient to consent to a particular procedure or treatment.Footnote 35
Informed consent is often considered part of tort law’s general requirement to act reasonably under the circumstances – a requirement that applies to physicians as well as manufacturers. Like the standard for manufacturers in negligence actions, the standard for physicians in negligence actions focuses on the reasonableness of the physician’s behavior. Unlike the standard of negligence for manufacturers, however, the standard of negligence for physicians is often determined by custom. What is reasonable, in other words, is determined by the jury based on what an actual doctor in that field of expertise would actually have done in the situation, rather than on what a reasonable doctor under the circumstances would have done.Footnote 36 This standard of care, however it is determined, applies to physicians who prescribe and use RPM devices. Thus, tort law will hold physicians liable if their failure to warn of device risks (if the learned intermediary doctrine applies) or to take reasonable care in monitoring or treating a patient, which can include inadequate training on how to use a device, causes harm to the patient.Footnote 37
C Defenses
Both physicians and manufacturers may have various defenses to claims involving defective products or negligent care. One is that the patient was negligent in using the device, and that negligence caused some or all of the harm suffered. In tort, a plaintiff’s negligence can affect his or her claims by (1) barring recovery entirely (contributory negligence), (2) reducing recovery by the percentage the plaintiff is at fault (pure comparative negligence), or (3) reducing recovery if the plaintiff’s fault is as great as or not greater than the defendant, otherwise barring recovery (modified comparative negligence). Most jurisdictions apply some version of modified comparative negligence when the plaintiff asserts a negligence claim. When the plaintiff asserts a claim in strict liability, contributory and comparative negligence defenses may still be available,Footnote 38 though they may be limited to certain evidentiary issues, such as risk-utility balancing or causation,Footnote 39 and circumscribed by statute.Footnote 40 Of course, even when comparative negligence applies, parceling liability may be challenging.
III Factors Affecting Liability Determinations
Building on the previous discussion, this part shows that how a device reaches the market and is used – through federal regulation, physician prescription, and patient and caregiver use – can also influence liability determinations.
A Regulation
How a device reaches the market can influence manufacturer liability for injuries caused by the device. RPM devices reach the market in two principal ways. New, high-risk devices (class III) must file a premarket notification approval (PMA) application that requires the manufacturer to demonstrate “reasonable assurance of the safety and effectiveness” of the device.Footnote 41 By contrast, if a manufacturer can justify that its device is “substantially equivalent” to a device already legally on the market, the device qualifies for clearance under section 510(k) of the Food, Drug, and Cosmetic Act (FDCA), an exception to the PMA process (class II).Footnote 42 Almost all devices that require premarket review enter the market through the 510(k) pathway, though the FDA does have the power to reclassify devices based on data showing novel risks.Footnote 43
Which of these two pathways applies to an RPM device can have important liability implications for the manufacturer and injured patient because the Supreme Court ruled that the Medical Device Amendments of 1976 (MDA) expressly or impliedly preempted state tort claims for high-risk devices that meet the “federal requirements” necessary for the approval of a PMA application.Footnote 44 Express preemption does not apply to devices cleared through the 510(k) pathway, which lacks the close regulatory review for safety and effectiveness present in a PMA review (Table 8.2).Footnote 45
Table 8.2 Express preemptive effect of MDA on tort claims, by defect alleged
| FDA review | Type of claim expressly preempted | ||
|---|---|---|---|
| Manufacture | Design | Marketing | |
| PMA | YesFootnote * | YesFootnote * | YesFootnote * |
| 510(k) | No | No | No |
| De NovoFootnote † | No (presumably) | No (presumably) | No (presumably) |
| None | No | No | No |
“Yes” means the claim is expressly preempted; “No” means the claim is not expressly preempted.
* Preemption does not bar parallel state claims.
† The de novo process has not yet been the subject of a preemption analysis. Given that it is designed to provide a 510(k)-like process for new devices, however, it is reasonable to assume that preemption analysis for devices authorized under the de novo review would be the same (or substantially the same) as those cleared through the 510(k) process. Courts analyzing the issue, however, may disagree with this assumption and make a contrary holding.
Implied preemption defeats only those parallel claims that would not exist but for the FDCA.Footnote 46 For 510(k)-devices, for example, implied preemption bars claims only when the manufacturer’s fraudulent representations caused the FDA to allow the marketing of a device it otherwise would not have (so-called state-law “fraud-on-the-FDA claims”) (Table 8.3).Footnote 47
Table 8.3 Express and implied preemptive effect of MDA on tort claims, by claim type
| FDA review | Preemption type | Type of claim preempted | ||
|---|---|---|---|---|
| Pathway | Express or Implied | Fraud-on-FDA | Parallel | Other State Law |
| PMA | Express and Implied | Yes | Some | Yes |
| 510(k) | Implied | Yes | No | No |
| De NovoFootnote † | Implied | Yes (presumably) | No (presumably) | No (presumably) |
| None | Implied | Yes | No | No |
“Yes” means the claim is expressly preempted; “No” means the claim is not expressly preempted. “(presumably)” means that courts would presumably find federal law impliedly preempted (or not) claims against manufacturers of devices authorized through the de novo pathway.
† The de novo process has not yet been the subject of a preemption analysis. Given that it is designed to provide a 510(k)-like process for new devices, however, it is reasonable to assume that preemption analysis for devices authorized under the de novo review would be the same (or substantially the same) as those cleared through the 510(k) process. Courts analyzing the issue, however, may disagree with this assumption and make a contrary holding.
As a result, a manufacturer’s liability exposure may turn on the type of product it manufactures and whether any similar product currently exists on the market. For example, if the heart rate monitoring feature of an implantable pacemaker is cleared through a 510(k) pathway, then the manufacturer would be liable for most harm that occurs as a result of a product defect.Footnote 48 If, by contrast, the feature required a PMA, then the manufacturer for which the PMA is granted would be immune from most lawsuits alleging injuries caused by the monitoring features of the device. Generally speaking, then, devices that undergo a more complete FDA review before market entry are subject to less tort liability than devices that undergo a less complete or no FDA review before market entry.
Consider the Sunrise Sleep Disorder Diagnostic Aid, which uses jaw movements to detect sleep apnea.Footnote 49 The device had no analogue on the market, but Sunrise filed to have its product cleared for the market without a PMA through an alternative mechanism, which may be treated similarly to the 510(k) process for preemption purposes.Footnote 50 While this choice likely saved Sunrise substantial capital, it could also increase its potential liability exposure. When deciding between a less stringent review and a PMA, Sunrise may have determined that the lower costs associated with less stringent review outweighed the benefits of liability protection afforded by a PMA.
Complicating things further, devices with a PMA are not immune from all lawsuits in all jurisdictions; such devices can be the subject of so-called “parallel claims” – state law causes of action that mirror FDA requirements but are not based solely upon them. For example, a state law manufacturing defect claim premised on, but not dependent on, a violation of federal manufacturing regulations could be a parallel claim provided that state law did not impose additional requirements on the manufacturer.Footnote 51 Here, jurisdictional issues can reappear because federal courts differ on what counts as a “parallel” claim that evades preemption.Footnote 52
B Path to Market and Patient
How an RPM device reaches the consumer can also influence physician and manufacturer liability. For example, Phillips manufactures the BioSticker System, which is an RPM device that attaches to the skin and measures physiological data, such as heart rate, respiratory rate, skin temperature, and other symptomatic or biometric data. This information is displayed on a dashboard that physicians can access and monitor.
The device, which was cleared under the 510(k) process,Footnote 53 originally required a physician’s prescription but, under a COVID-19 Emergency Use Authorization (EUA), is now available over the counter.Footnote 54 Before the EUA, this meant that the manufacturer could discharge its duty to warn by providing adequate instructions and warnings to the physician prescribing the device. The physician would then have an independent duty to obtain informed consent from the patient. After the EUA, however, consumers could access the device without a physician’s prescription, requiring that the warnings be made to the patient directly.
Because the learned intermediary doctrine affects manufacturer liability only for failure-to-warn claims, Phillips could still be liable for harm caused by manufacturing defects in the BioSticker System even prior to the EUA. Consider a situation in which, because of a manufacturing defect, the Biosticker device failed to transmit information to a physician showing an irregular heart rate and respiratory function. If the patient died as a result of the physician’s failure to intervene, and if the failure to intervene was caused by the device not having been manufactured according to specifications, then Phillips could be liable for the patient’s death.
Manufacturers may also be liable for some design defect claims even when the learned intermediary doctrine applies. The scope of this liability may depend on whether the device is prescribed by a physician and the type of device at issue. Phillip’s Biosticker was previously used by prescription, making it likely that Phillips could obtain immunity from strict liability design defect claims by adequately warning the physician of the risks posed by the device – for example, its inability to be used for more than a certain period of time or in water.Footnote 55
Once the FDA issued the EUA authorizing the device to be sold directly to consumers without a prescription, no amount of warning to physicians would likely insulate Phillips from strict liability design defect claims; however, in some, but by no means all, jurisdictions, adequately warning consumers may immunize manufacturers from design defect claims. A company like Empatica, for example, may try to immunize itself by warning physicians and consumers about the Embrace2’s ability to notify only emergency contacts, potentially foreclosing claims that Empatica defectively designed the Embrace2 since it lacked the capability to notify physicians or emergency responders. Regardless of whether strict liability immunity applies, a showing of adequate warning would not necessarily make Phillips immune from negligent design defect claims because of jurisdictional differences.
Besides the jurisdictional variations, it is unclear how courts would resolve such claims. While design defect claims often turn on the existence of available safer designs, along with the costs of developing and implementing them, some courts have been reluctant to apply this reasoning to prescription drugs.Footnote 56 Prescription RPM devices may be treated similarly. If they are not, however, such claims will turn on a fact-intensive analysis of the costs associated with changing the device to make it safer – rarely a question that can be resolved definitively and early in litigation.
In addition to its effect on manufacturer liability for information-based claims like failure to warn, the learned intermediary doctrine also opens physicians to more claims from patients injured by RPM devices. For example, suppose a physician prescribes to a patient, and the patient uses, a bracelet like the Embrace2 to detect seizure activity that automatically notifies designated caregivers.Footnote 57 If a seizure occurs and the device contacts a caregiver who cannot respond in time, the injured patient may attempt to sue the physician based on the theory that he or she would not have used the device if it was impossible for the device to alert someone who could more immediately help.Footnote 58
To avoid liability, physicians will need to properly inform and educate patients and caregivers about the devices’ risks and limitations. For devices like the Embrace2, part of this risk may be avoided by working with device manufacturers to notify parties who can respond in case of emergency and obtaining written and verbal consent, after explanation, for patient responsibilities in using the device and how the physicians can and will monitor the device data.
For example, physicians who recommend or prescribe a device like the Biosticker have a duty to understand how to use the product, including its limitations, as well as how and when they will be monitoring the data from the device. These physicians also have a duty to explain this clearly to the patient. If a physician will not be monitoring the device for real-time alerts, but instead using it as a data-gathering tool to obtain a more complete picture of the patient, they would do well to say so (and to document that conversation with the patient). The duty might include explaining to patients what to do if the device detects unusual behavior, including who they should contact and how they should interpret the data. Simply advising patients to “call 911” if there is an emergency may seem like a failsafe, but it also may create undue stress on the health care system if a device provides a variety of alerts. This may require new office procedures, points of contact, and protocols for reassessments of patients whose devices create particular kinds of alerts.
C Patient and Caregiver Use
Physicians are not the only individuals who can affect the liability of RPM device manufacturers. When patients use RPMs, they may be responsible for some or all of the harm the device causes, and their damages could be reduced or eliminated under the doctrine of comparative negligence. Similar to device manufacturers, how responsible patients are may turn on the type and nature of the device at issue.
Some RPM devices operate automatically and without any patient initiation, reducing the probability that a patient is responsible for harm suffered when using the device. RPM devices like the BioSticker or a pacemaker that monitors cardiovascular status, for example, collect information with minimal patient engagement. Without any patient action, it may be harder to show that the patient’s negligence, rather than the device, is the cause of any harm that occurred while the patient used the device.
However, other devices may require the patient to initiate, operate, or respond to them, and to do so under particular conditions or in a particular manner. For example, Google announced that it was developing a dermatology app that deploys artificial intelligence and machine learning to analyze user-uploaded photographs to track skin lesions over time and provide diagnostic information.Footnote 59 Hyfe, a smartphone app that likely will apply for 510(k) clearance,Footnote 60 uses similar technology to monitor cough data that the patient captures by affirmatively initiating the application. Patients who fail to track skin lesions at certain intervals using Google’s dermatology app or fail to initiate Hyfe may find that false negatives are their own fault, rather than the device’s. Moreover, patients who do not reasonably act on alerts from devices like RPMs may reduce or eliminate their ability to recover if they are injured as a result.
Patients could also see damages reduced or claims eliminated entirely when they use and rely on these devices in environments where manufacturers specifically state that they will not operate accurately. Thus, a patient who does not operate Hyfe or Google’s app in the recommended sound or lighting conditions, does not track coughs or skin lesions at the intervals required for the app to function optimally, or uses the device to predict asthma attacks or detect skin cancer (purposes for which they are not designed) may eliminate or reduce the probability of liability for manufacturers or physicians.
Similar issues apply to devices – like ResMed’s AirSense Elite 10 continuous positive airway pressure (CPAP) machine with built-in RPM – which not only treats sleep apnea, but also collects information about the person wearing it, that could be used to detect important health events, including a lack of oxygen being delivered to the user.Footnote 61 Patients who improperly place the mask on their face or use the device only sporadically will encounter challenges when suing manufacturers because a device did not detect a respiratory event. This may be true even if the device itself did not function properly.
Additionally, RPM devices may require manual patient data input to function properly. Medtronic offers a patient management system that uses both sensor-based RPM and self-reporting by patients to monitor and evaluate respiratory health, in particular patients with COVID-19.Footnote 62 Patients who enter information incorrectly may cause the system to incorrectly not recommend further care or alert the appropriate parties. If that happens and the patient is injured or dies as a result of the delay or absence of care, the patient may bear some or all of the responsibility for the harm, reducing or eliminating their recovery under the doctrine of comparative negligence.
Finally, third parties, like those who are “emergency contacts” alerted by a seizure detection device like the Embrace2, may have their phones turned off or may not respond to the patient in time to treat them. If their failure to respond causes harm to the patient, they could face liability, potentially reducing the liability of other actors. But if the third party’s inaction is caused by the patient’s failure to inform the third party that they would be notified, how they would be notified, or what they were expected to do when notified, then the patient may be responsible for the harm.
In short, the more patients can control what goes into the RPM, the more likely both the manufacturer and prescribing physician are to argue that any injury was caused not by them, but by the patient. To reduce the probability of patient-caused injury, manufacturers and physicians should carefully instruct patients on how, when, and for what purposes they should use RPM devices, and they should emphasize the limitations of the devices.
IV Conclusion
RPM devices may help patients self-manage conditions with fewer complications and at lower cost than traditional clinical care. But they also raise liability issues in tort law. While the doctrines used to assess these claims are quite old, their application to this new and changing area of medicine is unsettled. In this chapter, we have provided a framework for understanding these tort claims and how courts are likely to assess them based on a series of factors, including how the device reaches the market, the type of device, the type of claim, where it is brought, how it reaches the market and consumer, who uses it, and how they do so.
I Introduction
Health care’s digital transformation – accelerated, but by no means initiated, by the COVID-19 pandemic – has garnered attention as patients increasingly expect remote care options. A preponderance of digital health applications and connected sensors are poised to transform how health care is delivered in contexts outside of the hospital or clinic.Footnote 1
The digitization of health care delivery and medical technology raises questions about the safety of digital medical devices and how regulators monitor and respond to safety questions. One concern is that introducing software components to previously analog medical devices may create unexpected complexity or harm. For example, patients have died due to drug overdoses caused by “key bounce” in infusion pump software, whereby software incorrectly interprets a single keystroke as multiple keystrokes, resulting in patients receiving far more medicine than intended.Footnote 2
Even given the known safety concerns associated with digital products, the existing infrastructure for tracking medical device safety may not be well equipped to monitor the safety of products that are (increasingly) used outside of traditional health care facilities. Most post-market surveillance – that is, ongoing regulatory oversight beyond initial regulatory approval/clearance – in the United States takes the form of adverse event reporting by device manufacturers and (health care) user facilities or post-approval trials conducted by manufacturers.Footnote 3 Given that post-market surveillance primarily relies on the vigilance of manufacturers and health care providers, regulators may miss important safety signals as medical technologies are moved from health care facilities to patients’ homes.
These safety challenges have important implications for remote patient monitoring (RPM) tools. RPM is the collection of physiological measures that can be shared with health care providers – both actively by patients (e.g., by taking measurements and entering data at home) or passively with connected devices (which may automatically enter such data into a relevant database).Footnote 4 RPM encompasses the use of both combined hardware–software products, such as connected sensors, as well as standalone software tools.
Here, we focus specifically on the subset of RPM and other software-driven products that meet the definition of a medical device in the United States and, therefore, are subject to regulation by the US Food and Drug Administration (FDA). By focusing on regulated diagnostic and therapeutic devices, we specifically focus on products used in patients’ formal health care delivery instead of more consumer-health/wellness-oriented digital products. In other words, this chapter does not consider the overwhelmingly large set of consumer health apps that may or may not be verified or validated.Footnote 5 Importantly, we consider all medical devices containing software – both standalone software tools (often called “software as medical devices,” or SaMDs) as well as combination hardware–software products (“software in medical devices,” or SiMDs). In doing so, we follow the definition of “software-driven medical devices” (SdMDs) introduced by Gordon and Stern (2019) (which includes both SaMDs and SiMDs) and consider all SdMDs subject to FDA oversight.Footnote 6 Relative to digital diagnostics and therapeutics used outside of traditional clinical settings, our sample represents a highly relevant set of products, but is almost certainly a “super-set” of those regulated devices used in remote diagnosis and care.
The chapter proceeds as follows. First, we provide a brief overview of post-market surveillance of regulated medical devices in the United States and present data on post-market outcomes from recent years. Next, in detailed regulatory data, we identify SdMDs among regulated devices and document trends in their approvals, as well as the associated post-market safety issues. Finally, we conclude with a discussion of the implications of our findings for regulatory policy and the future of post-market surveillance for SdMDs.
II Post-Market Surveillance Activities and Regulatory Data
For regulated medical technologies, post-market surveillance plays an important role in ensuring that products continue to be safe and effective. The FDA’s Center for Devices and Radiological Health (CDRH) notes that post-market surveillance activities may include “tracking systems, reporting of device malfunctions, serious injuries or deaths, and registering the establishments where devices are produced or distributed.” Further, post-market requirements may also include surveillance studies and additional post-approval studies that were deemed to be required at the time of device approval.Footnote 7 We briefly summarize these activities and the types of publicly available data that they generate before turning to an empirical analysis.
Under 21 USC § 360I, the FDA has the authority to require manufacturers to engage in various post-market activities. These may be required at either the time of approval/clearance of a new device or sometime thereafter. An FDA Guidance Document further outlines best practices for the medical device industry with respect to several aspects of post-market surveillance,Footnote 8 including surveillance planning, interim reporting, and the implications of failing to comply with post-market reporting requirements. The following sections provide an overview of the various post-market activities that the FDA may require.
A Post-Market Trials and Registries
Two common ways in which manufacturers and regulators continue to monitor the ongoing safety and effectiveness of medical devices are via post-market clinical trials and patient registries.
One or more post-approval studies may be required by regulators at the time of a Pre-Market Approval (PMA), Humanitarian Device Exemption (HDE), or Product Development Protocol (PDP) application. The FDA may require that manufacturers commit to conducting such studies before it grants regulatory approval, and failure to complete studies may be grounds for the FDA to withdraw a device’s approval.Footnote 9 For example, the Post-Approval Study on Patients Who Received a HeartWare HVAD® During IDE Trials (HW-PAS-03), a multi-center study sponsored by the device’s manufacturer, provided continued evaluation and follow-up on patients who had received the HeartWare® Ventricular Assist System during earlier clinical trials.Footnote 10 The FDA may request that post-approval studies be conducted for both moderate- and high-risk devices. In practice, post-market studies are often delayed or terminated after the manufacturer changes the indication for use of the studied medical device.Footnote 11
Patient registries may be device-specific or embedded in larger surveillance initiatives. For example, as a condition for the approval of transcatheter heart valves, the FDA required all manufacturers to “continue to follow patients enrolled in their randomized studies for 10 years to further monitor transcatheter aortic valve safety and effectiveness….” As part of this initiative, the manufacturers agreed to participate in the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy (TVT) Registry.Footnote 12
B Plant Inspections
Another important component of post-market medical device regulation includes the inspection of plants where devices with hardware components are manufactured. Ball et al. (2017) summarized the rationale for manufacturing plant inspections by noting that “governments cannot feasibly sample every manufactured product before its release to customers; therefore, they frequently depend on plant inspections to appraise a plant’s quality systems.”Footnote 13
Generally speaking, device-manufacturing plant inspections are conducted according to the process described in the Quality System Inspection Technique Guide, which, in turn, follows the requirements contained within 21 CFR § 820.Footnote 14 Such plant inspections involve the detailed documentation of various processes – including those associated with quality system requirements, various forms of controls (e.g., design, production, and process), corrective and preventative actions, and so on. Notably, investigators do not inspect actual products, but, instead, examine the systems that guide the device manufacturing process.
Inspectors typically arrive at a plant unannounced, tour the facility, interview managers, and perform a process documentation review. There are three different types of such inspections: (1) Surveillance inspections – those that occur regularly and routinely to assess plant quality; (2) compliance inspections – those that are part of the establishment of new or modified manufacturing processes or new product launches; and (3) complaint inspections – those that occur in response to serious complaints by customers/device users.Footnote 15 In response to inspections, remedial actions may or may not be indicated; remedial actions may be “voluntary” or “official,” depending on the severity of issues identified.Footnote 16
C Medical Device Reporting
Once devices are legally marketed, a system of voluntary and mandatory medical device reporting serves to track adverse events and identify emergent safety issues. The FDA receives several hundred thousand medical device reports (MDRs) related to suspected device-associated malfunctions, injuries, and deaths annually.Footnote 17 These reports are collected in the Manufacturer and User Facility Device Experience (MAUDE) database, which is maintained by the FDA. Reports are mandatory for certain users – namely device manufacturers, importers, and health care facilities – and voluntary for others, including patients, consumers, and clinicians.
MDRs are input into the MAUDE database along with detailed product information, which includes a device’s manufacturer, product code, and FDA clearance/approval identifiers. This information allows individual MDRs to be linked to specific products. Although MDRs and the accompanying MAUDE database represent rich and well-organized sources of information, the FDA warns that the surveillance system may be incomplete, unverified, or inaccurate because of biased reporting, reporting lags, and other factors, and therefore cautions against using MAUDE data to understand the frequency or causality of adverse events. Nevertheless, MAUDE remains an important source of information about product quality issues, and its open-source format lends itself to empirical research in medicine and health policy.Footnote 18
D Recalls
Finally, post-market regulation includes the oversight of formal medical device recalls. Although recalls are typically manufacturer-initiated, they are overseen by the FDA, which classifies recalls according to risk/severity:
Class I recalls (most severe) occur where “there is a reasonable chance that a product will cause serious health problems or death” – for example, a faulty pacemaker lead that would prevent proper functioning.
Class II recalls (moderate severity) occur where “a product may cause a temporary or reversible health problem or where there is a slight chance that it will cause serious health problems or death” – for example, an insufficiently tight surgical clamp.
Class III recalls (low severity) occur where “a product is not likely to cause any health problem or injury” but where an issue nevertheless should be corrected – for example, a labeling issue.Footnote 19
The FDA’s medical device recall database publishes data on all classes of product recalls. The database links recall information to specific clearance/approval decision identifiers, enabling researchers to link a recall to at least one specific previously regulated product.
III Methods for Data Collection and Analysis
In this section we describe the datasets we used to quantify the likelihood of post-market safety events associated with SdMDs and other devices over recent years.
A Data Sources and Sample Construction
We identified all 510(k)-track and PMA-track medical devices (i.e., moderate and high-risk devices) cleared or approved by the FDA from 2008–2018 in the five common regulatory medical specialties (associated with CDRH Advisory Committees of the same name) most likely to include RPM devices: Cardiology, clinical chemistry, gastroenterology, general hospital, and general and plastic surgery. We then identified all recalls and adverse events associated with these devices that occurred between 2008 and 2020 using the FDA’s MAUDE and recall databases, respectively. We limited data from MAUDE to only include adverse events from mandatory reporters to reduce non-random differences in reporting across device types.
B Identifying Software-Driven Medical Devices
We employed a supervised document classification algorithm to identify SdMDs. For each medical device in our sample, we downloaded its associated public statement or summary document from the FDA’s website. These documents are required for all submissions and each “includes a description of the device such as might be found in the labeling or promotional material for the device.”Footnote 20 We then used optical character recognition software to search each document for the word “software” to identify devices with a software component.
This text search technique was demonstrated to work well in manual review: In comparison to a manually coded random sample of summary documents, the document classification had a 0 percent false negative rate, meaning devices flagged as including a software component via supervised document classification always included a software component. Accordingly, we identified a medical device as including a software component if “software” appeared at least once in its public summary of evidence. Additional details on the supervised document classification are provided elsewhere.Footnote 21
C Outcomes of Interest
We focused on two primary outcomes of interest: (1) Class I/II recalls (i.e., those of moderate or greater severity) and (2) mandatorily reported adverse events. For recalls, we identified all class I/II recalls that occurred within two years of regulatory approval/clearance for each device. We chose to use two years of follow-up, as most medical device recalls occur shortly after a medical device comes to market.Footnote 22 For adverse events, we similarly created a count of all adverse events from mandatory reporters in the two years following a device’s clearance/approval.
D Statistical Analysis
We compared differences in adverse events and recalls by software status by performing two-sided, two-sample t-tests comparing the outcomes between SdMDs vs. non-SdMDs. To understand the changes over time, we plotted the number of recalls or adverse events in a given calendar year divided by the number of approvals/clearances in the two preceding years, such that the frequency of outcomes was scaled by the number of devices recently placed on the market in each year. All statistical analyses were performed using data from the entire sample, as well as within individual medical specialties.
IV Results
Our sample included 13,186 medical devices, or 39.46 percent of all medical devices approved or cleared by the FDA during the sample period. During this time, software became increasingly prevalent in medical devices: While we observed variation over time in the total number and share of new SdMDs cleared/approved, all five medical specialties had a greater number and proportion of cleared/approved devices that included a software component in 2020 vs. 2010 (Figure 9.1). For example, 25.7 percent of the cardiovascular devices cleared or approved in 2010 included a software component, vs. 27.8 percent in 2020.
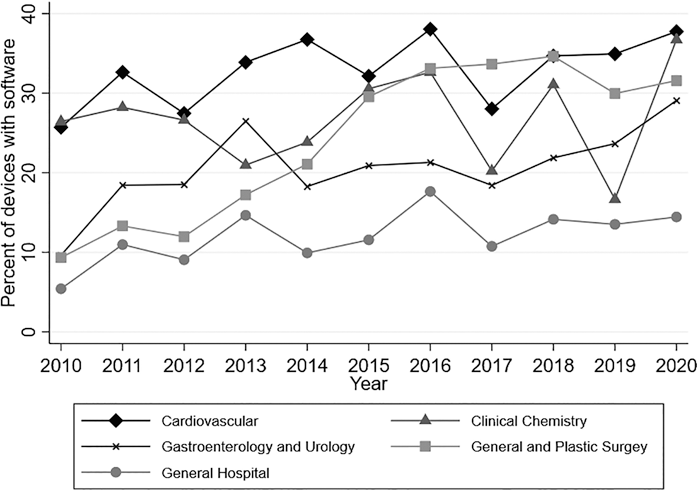
Figure 9.1 Proportion of devices with software by specialty over time
Note: Authors’ analysis of FDA approval and clearance data from 2010–2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of devices approved/cleared).
SdMDs in our sample experienced more adverse events (Figure 9.2) and class I/II recalls (Figure 9.3) than devices without software. The average SdMD had 14.516 associated adverse events from mandatory reporters in the MAUDE database (in its first two years on the market), while the average device without software had 3.524 associated adverse events reported (p = 0.010) (Table 9.1). Similarly, 8.1 percent of SdMDs experienced at least one class I/II recall in the two years following regulatory approval/clearance, vs. 3.6 percent of devices without software (p < 0.001) (Table 9.1).

A: No software.

B: Software.
Note: Authors’ analysis of FDA approval and clearance data and the FDA’s MAUDE database from 2010–2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each year–specialty observation, the total adverse events from mandatory reporters were calculated and then divided by the number of approvals and clearances within that specialty in the preceding two years.
Figure 9.2 Two-year adverse event rates by specialty over time.
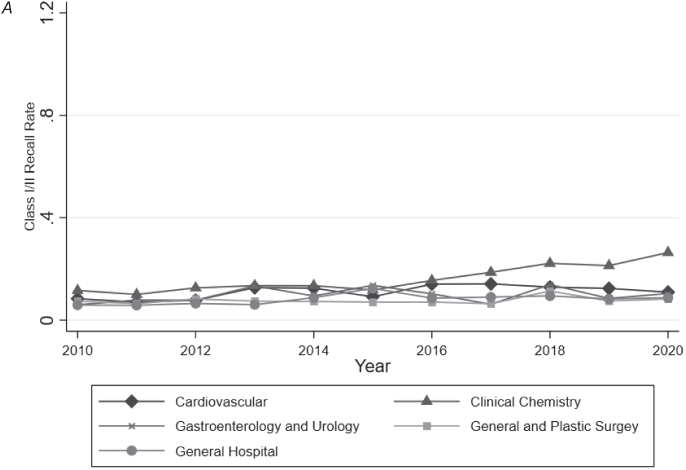
A: No software.

B: Software.
Note: Authors’ analysis of FDA approval and clearance data and the FDA’s MAUDE database from 2010 to 2020. Software identified based on keyword searches of FDA documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each year–specialty observation, total class I/II recalls were calculated and then divided by the number of approvals and clearances within that specialty in the preceding two years.
Figure 9.3 Two-year class I and class II recall rates by specialty over time.
Table 9.1 Two-year adverse event rates by specialty
| Specialty | Statistic | No software | Software | p |
|---|---|---|---|---|
| Cardiovascular | N | 3,055 | 1,341 | |
| Mean | 8.998 | 10.247 | 0.723 | |
| (SD) | (97.243) | (111.656) | ||
| Clinical chemistry | N | 1,067 | 332 | |
| Mean | 0.384 | 67.744 | 0.050 | |
| (SD) | (3.786) | (622.820) | ||
| Gastroenterology and urology | N | 1,530 | 329 | |
| Mean | 1.548 | 5.991 | 0.108 | |
| (SD) | (13.286) | (49.618) | ||
| General hospital | N | 2,214 | 263 | |
| Mean | 0.745 | 10.989 | 0.047 | |
| (SD) | (8.197) | (83.094) | ||
| General and plastic surgery | N | 2,424 | 631 | |
| Mean | 1.791 | 1.498 | 0.486 | |
| (SD) | (16.036) | (6.694) | ||
| Total | N | 10,290 | 2,896 | |
| Mean | 3.524 | 14.516 | 0.010 | |
| (SD) | (54.059) | (226.749) |
Note: Authors’ analysis of the FDA’s MAUDE and recall databases for devices approved/cleared from 2008 to 2018. Software identified based on keyword searches of FDA approval/clearance documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). Adverse events limited to mandatory reports. For each device, the total number of adverse events in two years following regulatory approval or clearance was calculated. Differences in means within specialties by software presence were assessed using two-sided t-tests under the assumption of unequal variance.
While devices with software generally experienced more adverse events and recalls, we observed significant heterogeneity in these differences by medical specialty area. When examining adverse events within individual medical specialties, only clinical chemistry and general hospital devices had statistically significant differences in adverse event rates in SdMDs vs. other devices. Among clinical chemistry devices, SdMDs had a mean 67.744 associated adverse events reported in the two years following regulatory approval or clearance, while non-SdMDs had a mean of just 0.384 adverse events reported in the two years following regulatory approval or clearance (p = 0.050) (Table 9.1). The difference between SdMDs and non-SdMDs, while statistically significant, was smaller among general hospital devices, where SdMDs had a mean of 10.989 associated adverse events in the two years following regulatory approval/clearance, while non-SdMDs had a mean of 0.745 adverse events reported over the same window of time (p = 0.047) (Table 9.1).
In contrast to adverse events, we observed significant differences in the number of recalls per approved device between SdMDs and non-SdMDs in each medical specialty studied. However, here too, the magnitude of the difference in recall rates varied meaningfully by specialty. General and plastic surgery devices had the smallest differences in recall rates (5.2 percent for SdMDs vs. 3.1 percent for non-SdMDs) (p = 0.025) (Table 9.2). General hospital devices had the largest difference in recall rates (11.8 percent of SdMDs vs. just 2.4 percent of non-SdMDs) (p < 0.001) (Table 9.2).
Table 9.2 Two-year class I and class II recall rates by specialty
| Specialty | Statistic | No software | Software | p |
|---|---|---|---|---|
| Cardiovascular | N | 3,055 | 1,341 | |
| Mean | 0.050 | 0.080 | <0.001 | |
| (SD) | (0.219) | (0.271) | ||
| Clinical chemistry | N | 1,067 | 332 | |
| Mean | 0.028 | 0.093 | <0.001 | |
| (SD) | (0.165) | (0.291) | ||
| Gastroenterology and urology | N | 1,530 | 329 | |
| Mean | 0.041 | 0.097 | 0.001 | |
| (SD) | (0.199) | (0.297) | ||
| General hospital | N | 2,214 | 263 | |
| Mean | 0.024 | 0.118 | <0.001 | |
| (SD) | (0.153) | (0.323) | ||
| General and plastic surgery | N | 2,424 | 631 | |
| Mean | 0.031 | 0.052 | 0.025 | |
| (SD) | (0.173) | (0.223) | ||
| Total | N | 10,290 | 2,896 | |
| Mean | 0.036 | 0.081 | <0.001 | |
| (SD) | (0.187) | (0.273) |
Note: Authors’ analysis of the FDA’s MAUDE and recall databases for devices approved/cleared from 2008 to 2018. Software identified based on keyword searches of FDA approval/clearance documents. Analysis restricted to medical specialties likely to include remote patient monitoring devices (39.46 percent of all devices approved/cleared). For each device, a binary indicator for a class I or class II recall was calculated. Differences in means within specialties by software presence were assessed using two-sided t-tests under the assumption of unequal variance.
We also observed that the differences in outcomes between SdMDs and non-SdMDs were driven in part by large increases in recalls and adverse events for specific types of devices over relatively short periods of time. For example, a large increase in recalls of general hospital devices between 2011 and 2013 was primarily driven by recalls of infusion pumps and sterilizers. A large increase in recalls of clinical chemistry devices in 2018 through 2020 was primarily driven by recalls of blood glucose monitors. Table 9.3 presents illustrative examples of such recalls.Footnote 23
Table 9.3 Example recalls
| Infusion pump recall description: | Glucose monitor recall description: |
|---|---|
| “Moog Inc. … announced today that the [FDA] has classified the voluntary correction of the Curlin 6000 CMS, Curlin 6000 CMS IOD, PainSmart, and PainSmart IOD as a Class I recall… The decision to conduct the device recall is due to a software anomaly which leads to software Error Code 45 (EC45), resulting in a shutdown of the pump. This failure may result in a delay or interruption of therapy, which could result in serious injury and/or death.” | “… Dexcom… issued a voluntary recall on the G6 CGM App due to the alarm feature on the iOS application failing to properly alert users. In particular, alarms were not detecting severe hypoglycemic (low glucose) or hyperglycemic (high glucose) events and therefore consumers were not being notified of fluctuations to blood glucose levels.” |
V Discussion
Overall, we observed that SdMDs had higher adverse event and recall probabilities compared to devices without software components. Further, we documented heterogeneity in the difference between SdMDs and non-SdMDs, both over time and across medical specialties.
It should be noted that there are several limitations on the current post-market surveillance system in the United States that prevent us from concluding that SdMDs are less safe than non-SdMDs. For example, even if SdMDs experience more recalls and adverse events, software-based recalls may have a smaller impact on patient wellbeing vs. other types of recalls. For example, manufacturers may be able to address (some) software recalls more quickly by issuing software patches, rather than physically removing defective products from the market. However, in supplemental analyses (not reported here), we found no evidence that recalls of SdMDs were terminated more quickly (on average) than those of non-SdMDs.
In addition to limitations in our ability to extrapolate patient impact from adverse event and recall-based measures, there is almost certainly imprecision in how we estimated the rates of these outcomes. The FDA’s MAUDE database for reporting adverse events does not include the number of devices in use at any given time – that is to say, there is no “denominator” to calculate the frequency of adverse events and/or recalls per device in circulation. As such, it is impossible to calculate a true adverse event rate, defined as adverse events per medical device in use. Rather, we calculate the rates of adverse events and recalls per device approved, but this is an imperfect measure. Devices with more units in circulation may have had more adverse events simply because they were used in more patients, which in turn, could impact the interpretation of our findings. Specifically, if SdMDs were used more (or less) frequently than non-SdMDs, the true per device used probability of such events could be substantially lower (or higher, respectively).
Further, both adverse event reporting and recalls rely on users and manufacturers identifying product problems. The salience of product issues is therefore likely to influence the probability with which true product failures are reported as adverse events and result in product recalls. One could imagine that certain types of product issues may be more noticeable in SdMDs – for example, issues with a digital display or internet connectivity. To the extent that this is true, it could also influence the results reported here and would drive up the likelihood that adverse events associated with SdMDs are reported and, as a corollary, the likelihood that a manufacturer recall is issued.
Our findings, therefore, also speak to the limitations of the current post-market surveillance and adverse event reporting infrastructure in the United States. While we found that on a per-new-device basis, SdMDs were more likely to experience recalls compared to non-SdMDs, we did not always detect differences in adverse events between SdMDs and non-SdMDs. Adverse events are a noisy signal of post-market safety and are not necessarily a reliable predictor of subsequent medical device recalls. The user-reported nature of the information collected in MAUDE may limit its ability to detect unsafe products, as regulators have already acknowledged.
Precisely because of these limitations, we believe that a key policy recommendation from our findings is the need for the systematic collection of unbiased data describing the post-market performance of both medical devices and digital diagnostics specifically. The FDA, the Centers for Medicare and Medicaid Services, and other bodies should work to include standardized medical device identifiers in administrative claims data (i.e., records of provider services reimbursed by health insurers).Footnote 24 Doing so would allow researchers and regulators to reliably track the use of SdMDs and their subsequent outcomes, thus differentiating safety issues from data artifacts caused by differences in device circulation.
It may also be beneficial for the FDA to consider implementing a broader and more robust set of post-market surveillance activities as software becomes increasingly integrated into medical devices and diagnostic technologies. Such activities could involve more direct evaluations of safety. For example, the FDA could potentially initiate periodic audits of randomly selected SdMDs to ensure that devices are performing as intended.
However, future post-market surveillance initiatives need not necessarily involve data collection by the FDA. The digitization of medical devices may raise safety issues, but it also presents new opportunities to collect data on device use and safety. SdMDs intrinsically generate “digital exhaust,” or metadata through their use. Regulators should consider how they might encourage manufacturers to leverage such data (including data on frequency and duration of device use) as part of post-market surveillance strategies, potentially by tying pre-market approval to a clear post-market data monitoring plan when appropriate.
The FDA alone will not be able to execute some of these changes. As the FDA acknowledged in a recent report, the “faster cycles of innovation and the speed of change for medical device software would benefit from a new regulatory approach,”Footnote 25 but the FDA is constrained in the actions it can currently take. The scope of the FDA’s regulatory activities is largely determined by the original 1976 legislation that gave the agency the authority to regulate devices. New legislative authority is needed for the FDA to design regulatory approaches that best address the unique nature of medical device software.Footnote 26
As the FDA considers new regulatory approaches to SdMDs, patients and providers should be aware that the introduction of software into previously analog devices may present new safety concerns. These concerns will not always be readily identifiable through existing post-market surveillance mechanisms. Accordingly, health care providers should consider how they might “monitor the monitors” and ensure that newly adopted remote patient monitoring technologies work as intended.
VI Conclusion
In an analysis focusing on five key medical specialties and using over a decade of data, we found that medical devices with software components had more adverse events and recalls (per new device) as compared to devices without software. While these findings hint at potential safety challenges associated with SdMDs, the data available do not allow us to extrapolate further and calculate safety issues per device in circulation, a measure that would be more appropriate for informing individual patient/provider safety concerns. That said, the data analyzed here demonstrate that it is vital to continue to monitor the safety and effectiveness of SdMDs going forward. Further, patients and providers should not assume that existing post-market surveillance mechanisms are sufficient for detecting safety concerns in the early years following market entry for new products with software components.
I Introduction
Artificial intelligence (AI), particularly its subcategory, machine learning, is changing our daily lives and the way we receive health care. The digital health apps market is booming, with over 350,000 health apps available to patients and consumers, ranging from wellness and fitness apps to disease management apps.Footnote 1 In particular, many direct-to-consumer medical AI apps for “self-diagnosis” (DTC medical self-diagnosing AI apps) are emerging that help individuals to identify a disease or other condition based on entering, for example, symptoms.Footnote 2 DTC medical self-diagnosing AI apps offer new opportunities, but they also raise issues. While the current legal debate has mainly focused on the poor accuracy of DTC medical self-diagnosing apps,Footnote 3 this chapter will discuss the labeling challenges associated with these apps that have received little attention in the literature.
This chapter will first explore the current and future landscape of DTC medical self-diagnosing AI apps. It will then focus on their regulation and discuss whether DTC medical self-diagnosing AI apps are medical devices under section 201(h)(1) of the Federal Food, Drug, and Cosmetic Act (FDCA). This will be followed by a discussion of two labeling challenges raised by DTC medical self-diagnosing AI apps: First, the concern of labeling DTC medical self-diagnosing AI apps as what I call “information-only” tools, and second, particular issues associated with the use of AI, ranging from bias to adaptive algorithms.
This chapter concludes that the labeling of DTC medical self-diagnosing AI apps as “information-only” rather than “diagnostic” tools is unknown to most consumers. The Food and Drug Administration (FDA) should create user-friendly labeling standards for AI-based medical devices, including those that are DTC. For example, these standards should ensure that consumers are adequately informed about the indications for use, model characteristics, and the risks and limitations of the respective DTC medical self-diagnosing AI apps. Based on a risk-based approach, some of these apps should also be prescribed by physicians rather than being offered directly to consumers over the counter. Physicians can help direct the use of the app in question and point out material facts, such as the risk of false positives and negatives, in the patient–physician conversation. In the long run, it may also be helpful to set up a new federal entity responsible for (at least for the coordination of) all issues raised by mobile health apps, from regulation to privacy to reimbursement. While this chapter focuses on FDA regulation for DTC medical self-diagnosing AI apps, some of the suggested solutions here may also have implications for other DTC apps.
II The Current and Future Landscape of DTC Medical Self-Diagnosing AI Apps
The US mobile health market is expected to grow continuously over the next decade, with medical apps (compared to fitness apps) representing the bulk of the market.Footnote 4 Before, or instead of, visiting a doctor’s office, consumers are trying more than ever before to self-diagnose their conditions by putting keywords of their symptoms into search engines like Google or using DTC medical self-diagnosing AI apps.Footnote 5 Approximately 80 percent of patients use the Internet for health-related searches.Footnote 6 According to a 2017 US survey, only 4 percent (ages 61 and older) to 10 percent (ages 18 to 29) of adults used apps for self-diagnosis, but 32 percent (ages 18 to 29) to 62 percent (ages 61 and older) of adults said that they could imagine using them.Footnote 7 Since the COVID-19 pandemic, digital health technologies have gained popularity to mitigate the spread of the virus,Footnote 8 and the use of medical self-diagnosing apps, including those based on AI, has become a reality for more adults in the USA.Footnote 9
In 2021, Google announced the planned launch of a pilot study of its “AI-powered dermatology tool” to help consumers find answers to their skin, nail, and hair condition questions.Footnote 10 With their phone’s camera, consumers simply need to take three photos of their skin, nail, or hair concerns from different perspectives and answer a few questions, such as their skin type and other symptoms.Footnote 11 The app will then offer a list of possible conditions.Footnote 12 Google’s app, dubbed DermAssist, is currently CE-marked as a low-risk (so-called class I) medical device in the European Union (EU) but is being further tested via a limited market release.Footnote 13 The CE marking indicates that the device conforms with the applicable legal requirements.Footnote 14 DermAssist is not yet available in the USA and has not undergone an FDA review for safety and effectiveness.Footnote 15
But Google is not the only company that is investing in dermatology apps. Indeed, a quick search in mobile app stores like Apple and Google Play reveals that there are already similar apps available to download for US consumers, such as AI Dermatologist: Skin Scanner by the developer Acina. Once consumers download this AI app, they can check their skin by taking a photo of, for example, their mole with their phone’s camera.Footnote 16 Within one minute, consumers will receive a risk assessment from AI Dermatologist, including some advice concerning the next steps.Footnote 17 It appears that AI Dermatologist is CE-marked as a medical device in the EU but has not undergone premarket review by the FDA.Footnote 18
There are also other DTC medical self-diagnosing AI apps already available on the US market. A classic example is Apple’s electrocardiogram (ECG) and irregular rhythms notification feature apps.Footnote 19 Both apps are moderate-risk (so-called class II) medical devices that received marketing authorization from the FDA in September 2018.Footnote 20 They are used with the Apple Watch and are addressed directly to consumers. While Apple’s ECG app is intended to store, create, transfer, record, and display a single channel ECG,Footnote 21 Apple’s irregular rhythms notification feature app detects irregular heart rhythm episodes suggestive of atrial fibrillation.Footnote 22 Another example is the AI symptom checker Ada. Consumers can manage their health by answering Ada’s health questions about their symptoms, such as headaches and stomach problems.Footnote 23 Ada’s AI will then use its medical dictionary of medical conditions and disorders to deliver possible causes for the symptoms and offer advice.Footnote 24 Ada’s consumer app is currently CE-marked as a class I medical device in the European Economic Area,Footnote 25 but, similar to AI Dermatologist, it does not appear that the app has undergone a premarket review by the FDA.Footnote 26
III DTC Medical Self-Diagnosing AI Apps as Medical Devices
Can the FDA regulate DTC medical self-diagnosing AI apps? The answer is yes, if they are classified as medical devices under FDCA section 201(h)(1). This section will discuss the definition of a medical device, the FDA’s enforcement discretion, and a relevant exception to the medical device definition.
A The Medical Device Definition and the FDA’s Enforcement Discretion
Under FDCA section 201(h)(1), a “device” is
an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is … intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man …, and which does not achieve its primary intended purposes through chemical action within or on the body of man … and which is not dependent upon being metabolized for the achievement of its primary intended purposes….Footnote 27
From the outset, the FDA can only regulate software functions that are classified as medical devices under FDCA section 201(h)(1) (so-called “device software functions”).Footnote 28 In other words, the FDA has no statutory authority to regulate software functions that are not considered medical devices under FDCA section 201(h)(1).Footnote 29 There are different types of software classifications. A relevant one is “Software as a Medical Device” (SaMD), which is standalone software and, as such, counts as a medical device.Footnote 30 The International Medical Device Regulators Forum defines SaMD as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”Footnote 31 For example, Apple’s ECG and irregular rhythms notification feature apps are both SaMD because they are software-only apps intended for a medical purpose.Footnote 32
Only recently, in September 2022, the FDA updated its Guidance for Device Software Functions and Mobile Medical Applications (Mobile Medical App Guidance) to reflect recent changes, such as the issuance of the FDA’s final Guidance on Clinical Decision Support Software.Footnote 33 Although the Mobile Medical App Guidance contains nonbinding recommendations, it represents the FDA’s current thinking on its regulatory approach to device software functions, including mobile medical apps.Footnote 34 The FDA defines “mobile medical apps” as mobile apps that incorporate device software functionalities that meet the medical device definition in the FDCA, and either are “intended … to be used as an accessory to a regulated medical device; or … to transform a mobile platform into a regulated medical device.”Footnote 35
The “intended use” is relevant for determining whether a mobile app is considered a medical device.Footnote 36 The term means “the objective intent of the persons legally responsible for the labeling of devices.”Footnote 37 Such persons are usually the manufacturers whose expressions determine the intent.Footnote 38 The intent can also be shown by the circumstances surrounding the product’s distribution.Footnote 39 For instance, the objective intent can be derived from advertising materials, labeling claims, and written or oral statements by the product’s manufacturer or its representatives.Footnote 40
In its Mobile Medical App Guidance, the FDA clarifies that it intends to focus its regulatory oversight on only those device software functions whose functionality could present a risk to the safety of patients if they were not to function as intended.Footnote 41 This means that the FDA intends to exercise enforcement discretion over those software functions that are or may be medical devices under FDCA section 201(h)(1) but present a lower risk to the public.Footnote 42 Enforcement discretion means that the agency does not aim to enforce requirements under the FDCA.Footnote 43
For example, the FDA intends to apply its regulatory oversight to device software functions that analyze images of skin lesions using mathematical algorithms and provide users with risk assessments of the lesions.Footnote 44 In contrast, for instance, the FDA considers apps exclusively intended for patient education, such as an app that helps guide patients to ask the right questions to their physician concerning their disease, as not being medical devices, and, thus, those apps fall outside of the FDA’s statutory authority.Footnote 45 An example of a mobile app that may meet the medical device definition, but for which the FDA intends to exercise enforcement discretion because it poses a lower risk to the public, is an app that provides a “Skill of the Day” behavioral technique that patients with diagnosed psychiatric conditions can access when experiencing increased anxiety.Footnote 46
When applying the FDA’s current thinking in the Mobile Medical App Guidance to DTC medical self-diagnosing AI apps, some of these apps are considered device software functions that are the focus of the agency’s regulatory oversight. Take as an example Apple’s ECG and irregular rhythms notification feature apps. Both apps are considered class II (moderate-risk) medical devices and had to undergo a premarket review by the FDA via the so-called De Novo process before being placed on the US market.Footnote 47
However, even if DTC medical self-diagnosing AI apps are considered medical devices because they help individuals identify a disease or other condition and are considered to be “intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease,”Footnote 48 the FDA may exercise enforcement discretion over some of them if they are considered to pose a low risk to the public. For example, as mentioned previously, the consumer app Ada is currently CE-marked as a class I (low-risk) medical device in the European Economic Area.Footnote 49 However, it seems that Ada has not undergone a premarket review by the FDA.Footnote 50 One option why this is likely the case is that Ada (may) meet(s) the medical device definition in FDCA section 201(h)(1),Footnote 51 but falls within the FDA’s enforcement discretion because it is considered to pose a lower risk to the public. This analysis also seems to be consistent with the Mobile Medical App Guidance. In Appendix B of its Guidance, the FDA lists examples of software functions that may meet the medical device definition but for which the agency exercises enforcement discretion, including:
– “Software functions that use a checklist of common signs and symptoms to provide a list of possible medical conditions and advice on when to consult a health care provider” and
– “Software functions that guide a user through a questionnaire of signs and symptoms to provide a recommendation for the type of health care facility most appropriate to their needs.”Footnote 52
In addition, most class I medical devices under the FDCA are also a priori exempt from premarket notification (510(k)) requirements.Footnote 53
B The Medical Device Exception, FDCA Section 520(o)(1)(B)
Section 3060(a) of the 21st Century Cures Act introduced five exceptions to the medical device definition for certain software functions. One of these exceptions is particularly relevant for DTC AI apps – namely FDCA section 520(o)(1)(B), which states that “the term device, as defined in section 201(h), shall not include a software function that is intended … for maintaining or encouraging a healthy lifestyle and is unrelated to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition; … .”
In 2019, the FDA issued nonbinding Guidance on Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act (Cures Act Guidance), in which the agency, among other things, expresses its current interpretation of FDCA section 520(o)(1)(B).Footnote 54 In particular, the FDA clarifies that FDCA section 520(o)(1)(B) means software functions that belong to the first category of general wellness intended uses, as defined in the FDA’s nonbinding Guidance on General Wellness: Policy for Low Risk Devices (General Wellness Guidance),Footnote 55 and that are “unrelated to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition.”Footnote 56 Software functions that fall within the first category of general wellness intended uses are intended for “maintaining or encouraging a general state of health or a healthy activity.”Footnote 57 For example, an app that assists users with weight loss goals and does not make any reference to diseases or conditions falls under FDCA section 520(o)(1)(B), and, thus, is not considered a medical device under FDCA section 201(h)(1).Footnote 58
In its Cures Act Guidance, the FDA also clarifies that software functions that fall within the second category of general wellness intended uses, as defined in the General Wellness Guidance, are not covered by FDCA section 520(o)(1)(B).Footnote 59 Software functions that fall within the second category of general wellness intended uses have “an intended use that relates the role of healthy lifestyle with helping to reduce the risk or impact of certain chronic diseases or conditions and where it is well understood and accepted that healthy lifestyle choices may play an important role in health outcomes for the disease or condition.”Footnote 60
In contrast to the first category of general wellness intended uses, this second category relates to the prevention or mitigation of a disease or condition, and, thus, software functions that fall within this second category are not excluded from the medical device definition.Footnote 61 For example, if the app in the previous example makes reference to diseases or conditions – for instance, if it claims that maintaining a healthy weight will aid living well with type 2 diabetes – this app falls outside of the scope of FDCA section 520(o)(1)(B).Footnote 62
As understood here, DTC medical self-diagnosing AI apps help users to identify a disease or other condition based on entering, for example, symptoms. They are related “to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition” and, thus, do not fall under the medical device exception in FDCA section 520(o)(1)(B).Footnote 63 To sum up, DTC medical self-diagnosing AI apps are medical devices under FDCA Section 201(h)(1) that are either the focus of the FDA’s regulatory oversight or for which the agency exercises its enforcement discretion. Figure 10.1 summarizes the regulation of mobile health apps, including DTC medical self-diagnosing AI apps.

Figure 10.1 Regulation of mobile health apps, including DTC medical self-diagnosing AI appsa
aFigure inspired by the FDA’s Mobile Medical App Guidance, supra note 33; the FDA’s Cures Act Guidance, supra note 54; the FDA’s General Wellness Guidance, supra note 55.
IV Labeling Challenges for DTC Medical Self-Diagnosing AI Apps
As established above, DTC medical self-diagnosing AI apps, as understood here, are medical devices that are either the focus of the FDA’s regulatory oversight or for which the agency exercises its enforcement discretion. This section will focus on the labeling challenges for DTC medical self-diagnosing AI apps. It will first give an overview of medical device labeling and the relevant terminology. It will then focus on labeling challenges for DTC medical self-diagnosing AI apps and make suggestions on how to overcome them.
A Labeling
Device software functions are organized into one of three classes based on their risk level, ranging from class I (lowest risk) to class III (highest risk).Footnote 64 Depending on the device classification, manufacturers must follow the associated controls – that is, General Controls, Special Controls, and/or Premarket Approval.Footnote 65 In principle, General Controls apply to all device software functions.Footnote 66 For instance, the General Device Labeling Requirements in Part 801 of Title 21 of the Code of Federal Regulations (CFR) are General Controls.Footnote 67 21 CFR Part 801 includes, among other things, general labeling provisions, such as the name and place of business, adequate directions for use, and the use of symbols, as well as special requirements for specific devices, such as hearing aids, and labeling requirements for unique device identification and over-the-counter devices.Footnote 68
Labeling is defined in FDCA section 201(m) as “all labels and other written, printed, or graphic matter (1) upon any article or any of its containers or wrappers, or (2) accompanying such article.” It is a generic term that also includes all labels.Footnote 69 Under FDCA section 201(k), the term “label” means
a display of written, printed, or graphic matter upon the immediate container of any article; and a requirement made by or under authority of this Act that any word, statement, or other information appear on the label shall not be considered to be complied with unless such word, statement, or other information also appears on the outside container or wrapper, if any there be, of the retail package of such article, or is easily legible through the outside container or wrapper.
In the context of DTC medical self-diagnosing AI apps, the label will usually be available in non-physical form through the app itself.
It is also worth noting that if the “labeling is false or misleading in any particular,” the device is considered misbranded under FDCA section 502(a)(1). The term “misleading” means that the labeling proves deceptive to device users and creates or leads to a false impression in their minds.Footnote 70 For example, this can be the case if the label contains exaggerated claims or if it fails to inform users about relevant facts.Footnote 71
B Challenges
DTC medical self-diagnosing AI apps raise labeling challenges. This section will discuss two: First, the concern of labeling DTC medical self-diagnosing AI apps as what I call “information-only” tools, and second, particular issues associated with the use of AI, ranging from bias to adaptive algorithms. It will also make suggestions on how to address these challenges. While the following remarks focus on medical devices, they may also have implications for those DTC apps that fall outside the FDA’s statutory authority.
i Labeling as “Information-Only” Tools
Apple’s ECG and irregular rhythms notification feature apps used with the Apple Watch are both over-the-counter class II medical devices that received marketing authorization from the FDA in September 2018.Footnote 72 As previously mentioned, Apple’s ECG app is intended to store, create, transfer, record, and display a single channel ECG.Footnote 73 The indications for use, however, also include, among other things, the following sentences:
The user is not intended to interpret or take clinical action based on the device output without consultation of a qualified healthcare professional. The ECG waveform is meant to supplement rhythm classification for the purposes of discriminating AFib [atrial fibrillation] from normal sinus rhythm and not intended to replace traditional methods of diagnosis or treatment.Footnote 74
The FDA created a new device type, namely “electrocardiograph software for over-the-counter use,” regulated in 21 CFR 870.2345, for Apple’s ECG app and substantially equivalent devices.Footnote 75 Interestingly, 21 CFR 870.2345(a) also states that “this device is not intended to provide a diagnosis.”
Moreover, as mentioned, Apple’s irregular rhythms notification feature app detects irregular heart rhythm episodes suggestive of atrial fibrillation.Footnote 76 But, much like Apple’s ECG app, this app’s indications for use include, inter alia, the following phrases:
It is not intended to provide a notification on every episode of irregular rhythm suggestive of AFib and the absence of a notification is not intended to indicate no disease process is present; rather the feature is intended to opportunistically surface a notification of possible AFib when sufficient data are available for analysis. These data are only captured when the user is still. Along with the user’s risk factors, the feature can be used to supplement the decision for AFib screening. The feature is not intended to replace traditional methods of diagnosis or treatment.Footnote 77
The FDA also created a new device type, namely “photoplethysmograph analysis software for over-the-counter use,” laid down in 21 CFR 870.2790, for Apple’s irregular rhythms notification feature app and substantially equivalent devices.Footnote 78 Similar to 21 CFR 870.2345, this regulation also clarifies that “this device is not intended to provide a diagnosis.”Footnote 79
But Apple’s apps are not the only DTC medical self-diagnosing AI apps that articulate that their device “is not intended to provide a diagnosis.” For example, Google’s 2021 announcement of its AI-powered dermatology tool says:Footnote 80 “The tool is not intended to provide a diagnosis nor be a substitute for medical advice as many conditions require clinician review, in-person examination, or additional testing like a biopsy. Rather we hope it gives you access to authoritative information so you can make a more informed decision about your next step.”Footnote 81
In addition, Google’s website states: “DermAssist is intended for informational purposes only and does not provide a medical diagnosis.”Footnote 82 The same is also true for the AI Dermatologist: Skin Scanner app.Footnote 83 When looking up information about the app in an app store, the preview states: “It is essential to understand that an AI-Dermatologist is not a diagnostic tool and cannot replace or substitute a visit to your doctor.”Footnote 84 App store previews of Ada say something similar: “CAUTION: The Ada app cannot give you a medical diagnosis…. The Ada app does not replace your healthcare professional’s advice or an appointment with your doctor.”Footnote 85
Consequently, DTC medical self-diagnosing AI apps are labeled as “information-only” rather than “diagnostic” tools.Footnote 86 Irrespective of whether DTC medical self-diagnosing AI apps are medical devices that are the focus of the FDA’s regulatory oversight or for which the agency exercises its enforcement discretion, these apps seem to have in common that their manufacturers claim they are “not intended to provide a diagnosis.” This is likely due to their over-the-counter nature, although Apple’s clinical study of the ECG app, for example, showed that the app correctly diagnosed atrial fibrillation with 98.3 percent sensitivity and 99.6 percent specificity.Footnote 87 As a comparison, a prescription device is a “device which, because of any potentiality for harmful effect, or the method of its use, or the collateral measures necessary to its use is not safe except under the supervision of a practitioner licensed by law to direct the use of such device.”Footnote 88 But do patients and consumers really understand that Apple’s ECG app and similar apps are not intended to replace traditional diagnosis and treatment methods, let alone that some have been FDA reviewed and others have not?
There appears to be a significant discrepancy between the user’s perception of the intended use of DTC medical self-diagnosing AI apps and their actual intended use (i.e., not to diagnose). Indeed, a recent study on AI-assisted symptom checkers revealed that 84.1 percent of respondents perceive them as diagnostic tools.Footnote 89 In addition, 33.2 percent of respondents use symptom checkers for deciding whether to seek care, and 15.8 percent of respondents said they use them to receive medical advice without seeing a doctor.Footnote 90 However, as seen above, apps like the ones from Apple and other companies have clear indications for use, and, thus, are likely not considered deceptive to device users and, thus, not “misleading” under FDCA section 502(a)(1).Footnote 91 Nevertheless, even if one cannot establish misleading labeling under the FDCA, there is this misperception among users that these apps are diagnostic tools.
This misperception can also be due, among other things, to the fact that many users may not read the labels. Labeling has many benefits, including helping patients and consumers to make more informed decisions, such as by informing them about the potential limitations of an app. But if users do not read the labels and accompanying statements and language like “this device is not intended to provide a diagnosis” is buried somewhere within them, using DTC medical self-diagnosing AI apps can become risky and jeopardize patient health. For example, imagine a patient uses an app like AI Dermatologist and screens herself for skin cancer. What if the AI misses a melanoma, and the patient does not see a doctor because she perceives the app as a diagnostic tool and believes everything is alright?
Regulators and stakeholders, such as app developers, need to better educate users of DTC medical self-diagnosing AI apps, for example, about the indications for use, whether the app has undergone FDA review, and its risks. With the right design, labels could help to achieve these goals. Several groups have already shown the benefits of “eye-popping” label designs, such as with the help of “nutrition” or “model facts” labels.Footnote 92 In particular for apps, there is a multitude of possible design options (e.g., pop-up notifications in plain language) to make users more effectively aware of important information.Footnote 93 Thus, regulators like the FDA could – with the help of stakeholders and label designers – develop user-friendly label design options for DTC medical self-diagnosing AI apps.Footnote 94 Once created, additional educational campaigns could be used to promote the proper reading of the label.Footnote 95 Human factors testing would also be helpful, particularly to see whether users understand when to seek medical help.Footnote 96
In addition, as part of its regulatory review, the FDA should consider whether some of these apps should be prescribed by doctors rather than being offered directly to consumers over the counter.Footnote 97 The advantage could be that physicians could assist patients with the use of the app in question and point out material facts in the patient–physician conversation. A risk-based approach may likely be useful here to determine such “prescription apps.”
Moreover, there is a general question of whether the FDA’s current approach to practice enforcement discretion over many DTC medical self-diagnosing AI apps is convincing. Other countries have come up with different regulatory designs to better protect consumers. For example, Germany incentivizes manufacturers of even low-risk apps (i.e., class I devices) to produce high-quality apps that comply with specific standards (e.g., safety, privacy, etc.) by offering insurance coverage for their apps in return.Footnote 98 While the FDA does not currently seem to have the resources to execute a similar approach and review all DTC medical self-diagnosing AI apps, the flood of mobile health apps and all the associated issues,Footnote 99 ranging from poorly designed products to inadequate data protection, to labeling issues and misperceptions concerning their use, requires a new regulatory approach in the long run. A better option might be to create a new federal entity in the future that would be responsible for (at least the coordination of) all issues raised by mobile health apps, including DTC medical self-diagnosing AI apps, from regulation over privacy to enforcement actions and reimbursement.
ii Particular Issues of AI: From Bias to Adaptive Algorithms
Another labeling challenge that DTC medical self-diagnosing AI apps raise is that they are not only directly addressed to consumers without a licensed practitioner’s supervision, but that they also operate using AI. Indeed, AI-based medical devices, including DTC medical self-diagnosing AI apps, are very different from traditional medical devices, such as simple tongue depressors.Footnote 100
First, DTC medical self-diagnosing AI apps may use methods like deep learning that make them opaque (often dubbed “black boxes”).Footnote 101 This means that the end users of the DTC medical self-diagnosing AI app (and likely even the app developers) cannot understand how the AI reached its recommendations and/or decisions. Second, DTC medical self-diagnosing AI apps may be biased. AI tools are prone to different types of bias, ranging from biased data fed to them (e.g., a skin cancer screening app that is largely trained on white skin images) to label choice biases (e.g., the algorithm uses an ineffective proxy for ground truth).Footnote 102 Third, DTC medical self-diagnosing AI apps may continuously learn from new data (e.g., health information, images, etc.) supplied by consumers using such apps (so-called “adaptive algorithms”).Footnote 103 These apps are, thus, much more unpredictable in terms of their reliability and would preferably need constant monitoring to avoid introducing new biases, for example.Footnote 104 Lastly, the human–AI interaction is complex. In particular, DTC medical self-diagnosing AI apps that have unique characteristics as their outputs are often probabilistic and, thus, require consumers to incorporate the information received into their own beliefs.Footnote 105 In addition, DTC medical self-diagnosing AI apps are usually available for little money or even for free.Footnote 106 They can easily be used as often as consumers wish.Footnote 107 For example, consumers of a skin scanner app may decide to scan their moles many times (rather than just once), which increases the chance of false-positive results – that is, the app detects a potential disease that is not actually present.Footnote 108 Because consumers are typically risk-averse about their health outcomes, they may seek medical help when it is not needed, further overburdening the health care system and taking away limited resources from patients who are more likely to need them.Footnote 109
Despite the differences between AI-based medical devices, such as DTC medical self-diagnosing AI apps, and traditional medical devices, such as simple tongue depressors, there are currently no labeling requirements for medical devices specifically aimed at AI (see Title 21 of the CFR).Footnote 110 The FDA has not yet developed any labeling standards for AI-based medical devices, let alone those directly addressed to consumers.Footnote 111 Thus, when creating the optimal design labels for DTC medical self-diagnosing AI apps,Footnote 112 the FDA should also focus on the content and use this opportunity to develop labeling standards for AI-based medical devices, including those that are DTC.Footnote 113
It is crucial that consumers know and understand, among other things, the indications for use, model characteristics, and the risks and limitations of AI-based medical devices.Footnote 114 For example, users of DTC medical self-diagnosing AI apps should be made aware of the type of AI used (e.g., a black box, an adaptive algorithm, etc.) and the risks associated with using the app in question. They should also be informed about the various risks of bias and warned against blindly relying on the app’s outputs. Moreover, consumers should be alerted to the fact that increased testing can lead to an increased chance of false positives and generally be educated about the risks of false-positive and false-negative results, including when to see a doctor. A discussion with stakeholders needs to occur as soon as possible on the content of the labels of AI-based medical devices, including DTC medical self-diagnosing AI apps.Footnote 115 In particular, the language used for the labeling of these devices will need to be plain when they are DTC.Footnote 116
V Conclusion
The digital health apps market is booming, and DTC medical self-diagnosing AI apps are emerging that help users to identify a disease or other condition based on entering, for instance, symptoms. Examples of such apps include Apple’s ECG and irregular rhythm notification feature apps, Google’s AI-powered dermatology tool, the AI Dermatologist: Skin Scanner app, and the symptom checker Ada. DTC medical self-diagnosing AI apps raise a multitude of challenges, including questions of labeling. What should labels directly addressed to consumers look like? What information should be included in such a label?
This chapter has argued that the FDA should develop user-friendly labeling standards for AI-based medical devices, including DTC medical self-diagnosing AI apps. For example, consumers need to be effectively informed about the type of AI used (e.g., a black box, an adaptive algorithm, etc.), the various risks of bias, the risks of false-positive and negative results, and when to seek medical help. In particular, the design of such labels needs to promote their reading so that users are made aware that the DTC medical self-diagnosing AI app in question is an “information-only” tool and is “not intended to provide a diagnosis.” Additionally, some of these apps should be prescribed by a doctor, not offered over the counter, based on a risk-based approach so that the doctor can point out key facts. In the long run, it may also be helpful to create a new federal entity responsible for (at least the coordination of) all issues raised by mobile health apps, ranging from regulation to privacy to reimbursement.
I Introduction
Digital technologies are integrated into all areas of life. The field of health is no exception. Some of the earliest uses of digital technology for health can be dated back to the 1960s.Footnote 1 In its 2005 resolution, the World Health Assembly (WHA) acknowledged the value of digital health and encouraged its member states to incorporate digital technologies into their health systems.Footnote 2 The important role of digital health was reiterated in the 2018 resolution, in which the WHA urged member states to prioritize the development and greater use of digital technologies for promoting equitable, affordable, and universal access to health for all.Footnote 3 During the COVID-19 pandemic, many countries have accelerated the utilization and development of digital health so as to guarantee the continued provision of health services with minimum in-person contact. As a result, there is now a growing consensus among countries that digital health has the potential to strengthen health systems and improve access to health.Footnote 4
China embraced the new digital technology and attempted to use it for health as early as the 1990s.Footnote 5 As will be discussed in the following sections, the government, encouraged by the rapid development of internet technology in China, has made great efforts to support digital health in the past three decades for solving the problem of uneven geographic and health resources distribution. In 2018, the General Office of the State Council released an overarching document, entitled Opinions on Promoting the Development of “Internet Plus Health Care,” with an aim to promote the innovative integration of digital technologies into the health system as a means of improving equitable, affordable, and universal access to health.Footnote 6 The term “internet plus health care” (IPHC) was introduced as a blanket term to mean the use of digital technologies in support of the delivery of health care and health-related services, such as internet-based diagnosis, treatment, and medicine, and internet hospitals. In this article, we use IPHC as an umbrella term for general discussion and refer to specific terms such as internet-based diagnosis where necessary.
This article intends to provide an overview of the development of IPHC in China, from its origins to its widespread use during the COVID-19 pandemic, with focuses on its regulatory landscape and, particularly, on digital diagnosis. In Section III, we identify three major regulatory challenges to IPHC. We conclude with a few recommendations for furthering the development and implementation of IPHC in the post-COVID-19 era.
II Landscape Analysis of “Internet Plus Health Care”
A The Development of “Internet Plus Health Care” in China
China’s health system has long been criticized for its inequitable distribution of health resources and unequal access to health care. To address these deeply rooted problems, particularly the weak provision of primary health care at grassroots level, the Chinese central government initiated a new round of health reform in 2009. Digital technologies, across a range of measures, have been employed as a feasible modern channel for promoting equitable, affordable, and universal access to health for all.Footnote 7
As far back as the 1990s, some of the first attempts at using digital technologies to improve access to quality health services were initiated. In 1988, the first remote consultation center was founded, which enabled the discussion of neurosurgery cases between Chinese and German hospitals via satellite.Footnote 8 With the development of information technology (IT), many medical institutions in urban areas started to establish remote consultation centers for exchanging knowledge and sharing experience with lower-level medical institutions. More importantly, the government made special efforts to support remote diagnosis in rural and mountainous regions as a means of addressing geographic barriers to access health care services. For example, many village clinics were equipped with computer terminals, despite the then poor IT infrastructure in these regions. As a result, a relatively robust physical and IT infrastructure was deployed for IPHC.
Since the beginning of the twenty-first century, a variety of regulatory and policy instruments have been adopted to facilitate the development of IPHC. With supportive policies, giant IT companies such as Alibaba and Tencent began to leverage their advances in digital technologies to establish online platforms and mobile applications to provide health-related services. In the meantime, public medical institutions also started to establish their own internet platforms. In 2012, the first public online hospital platform was founded in Guangdong Province.Footnote 9 Provinces with scarce health resources took the initiative to issue favorable policies to attract medical companies to set up internet hospitals as a means of improving access to health for their residents. The favorable policies and innovative technologies have stimulated the rapid development of IPHC during this period. In 2018, the aforementioned Opinions on Promoting the Development of “Internet Plus Health Care” (Opinions) document was released, with an overall aim to promote IPHC and guarantee equitable, affordable, and universal access to health for all. For quality assurance purposes, platform-based internet hospitals with no offline facilities were no longer allowed. In particular, this document required authorities to develop implementation rules and action plans for governing IPHC. A preliminary regulatory framework was thus established (see details in Section II.B). In September 2018, the National Health Commission (NHC) and the Government of Ningxia Hui Autonomous Region signed a strategic agreement to establish the first national IPHC pilot demonstration area, and in May 2019, the NHC signed similar agreements with another ten provinces and municipalities. In short, tremendous efforts had been made to promote the development and use of IPHC before the COVID-19 pandemic. However, the use of IPHC remained limited in practice due to regulatory restrictions and poor technical maintenance.
The COVID-19 outbreak has become a turning point in this area. The Chinese government has made several regulatory changes to make IPHC more widely used and to ensure the continued provision of health care when in-person services were not available during the health emergency. These changes include the relaxation of limitations on the scope of IPHC services and the expansion of health insurance coverage. The NHC also issued guidelines urging public hospitals to introduce or further develop IPHC as a means of relieving pressure on overloaded offline facilities. As a result, IPHC has obtained greater acceptance and its use surged during the pandemic. Statistical reports show that, by 2021, the number of licensed internet hospitals in China exceeded 1,600, while the user size of IPHC amounted to 298 million, accounting for 28.9 percent of all Internet users.Footnote 10
B Current Regulatory Framework of “Internet Plus Health Care”
In 2018, the NHC and National Administration of Traditional Chinese Medicine (NATCM) issued three consecutive normative documents for trial implementation as a response to the requirements of the Opinions mentioned above: The Administrative Measures for Internet-based Diagnosis and Treatment (AMIDT), Administrative Measures for Internet Hospital (AMIH), and Administrative Regulations on Remote Medical Service (ARRMS).Footnote 11 The AMIDT and ARRMS provide norms and guidelines for the provision of “internet-based diagnosis and treatment” and “remote diagnosis and treatment.”Footnote 12 These two documents also make it clear that medical institutions and qualified health personnel are eligible to provide such services. According to the AMIH, there are two different operating models of internet hospitals. The AMIH stipulates stringent licensing and operation requirements for each type of internet hospital. It also sets out registration and practicing requirements for physicians who practice at internet hospitals.
In addition, as will be discussed further in Section III.C, the National Health Security Administration (NHSA) issued a series of guidance documents regarding the reimbursement and coverage of internet-based medical services during the pandemic, so as to make IPHC more widely affordable to patients.
Safety is at the heart of health care services, and internet-based diagnoses are no exception. After three years of trial implementation, the NHC published its Regulatory Rules on Internet-based Diagnosis and Treatment in March 2022, with an aim to reinforce governance structures and oversight mechanisms for internet-based diagnosis as well as the related medical institutions and health personnel.Footnote 13 The new Regulatory Rules set out guiding principles for the supervision of internet-based diagnosis and outlined explicit regulatory requirements for medical institutions providing such services. This regulatory document requires provincial health administrations to establish their own regulatory platforms and implement real-time supervision of medical institutions that provide internet-based diagnosis within their jurisdiction, to ensure that internet-based diagnoses meet the same quality as in-person services. Built on these documents, a preliminary regulatory framework for IPHC has been created. Table 11.1 summarizes the legal and policy documents that have an impact on IPHC.
Table 11.1 A selection of legal and policy documents that impact IPHC
| 2012 | Administrative Measures for Remote Medical Care (for Trial Implementation) |
| 2014 | Opinions on Promoting Medical Institutes’ Delivery of Remote Medical Services |
| 2015 | Guiding Opinions of the State Council on Actively Advancing the “Internet Plus Action” |
| 2016 | “Healthy China 2030” Plan |
| 2017 | Administrative Regulations on the Application of Electronic Medical Records (for Trial Implementation) |
| 2018 | Administrative Measures for Internet-based Diagnosis and Treatment (for Trial Implementation) Administrative Measures for Internet Hospital (for Trial Implementation) Administrative Measures on the Standards, Security and Services of National Healthcare Big Data (for Trial Implementation) Administrative Regulations on Remote Medical Service (for Trial Implementation) Opinions on Promoting the Development of “Internet Plus Health Care” |
| 2019 | Basic Medical and Health Care and Health Promotion Law Guiding Opinions on Improving the “Internet Plus” Medical Service Price and Medical Insurance Coverage Policy |
| 2020 | Guiding Opinions on Actively Promoting Medical Insurance Coverage of “Internet Plus” Medical Service Guiding Opinions on Promoting “Internet Plus” Medical Insurance Service during the Prevention and Control of COVID-19 Information Security Technology-Guide for Health Data Security (GB/T 39725-2020) |
| 2022 | Law on Physicians Regulatory Rules on Internet-based Diagnosis and Treatment (for Trial Implementation) |
C Types of “Internet Plus Health Care” Services
i Internet-Based Diagnosis
Internet-based diagnosis, or online diagnosis, is a particular type of medical service precisely defined by the AMIDT as “a follow-up diagnosis for some common and chronic diseases delivered by a medical institution’s own registered physicians via internet or other digital technologies.”
Several restrictions in the AMIDT have been imposed on internet-based diagnosis for quality assurance purposes. First, only medical institutions with valid licenses and registered physicians with more than three years of independent clinical practice are qualified to provide internet-based diagnoses. Second, the scope of diseases is limited to certain common and chronic diseases. The types of chronic disease are determined by provincial health commissions and health security administrations, and generally include hypertension, coronary heart disease, diabetes, epilepsy, and so on. Third, a first diagnosis or diagnoses of sophisticated diseases are not permitted. This means that if a person becomes ill and in need of medical services, the person has to have a face-to-face diagnosis first. A physician in an offline hospital should diagnose that the patient has a common or chronic disease, then follow-up diagnoses and treatment can be given online. First diagnoses or patients with no medical records are not eligible for an internet-based diagnosis. Requiring an in-person diagnosis for a first diagnosis is a particular procedural and institutional requirement for safety assurance in the field of digital health.
ii Remote Diagnosis
In the Chinese context, remote diagnosis is a type of medical service provided by two or more medical institutions that are generally in the same medical consortium. According to the ARRMS, one medical institution can invite another to provide technical support for the diagnosis of its patients by means of digital technologies. In practice, normally the inviter is a community-level medical institution that has a close partnership (e.g., medical consortium) with the invitee, which, in most cases, is a top-tier medical institution. The invited medical institution will provide remote diagnosis on the basis of physical examinations and diagnostic tests, such as X-ray, ultrasound, and electrocardiogram, conducted by the inviting institution. For example, a township-level medical center may be equipped with an X-ray unit but lack the expertise to diagnose on the basis of an X-ray film. If a person living in this kind of rural area breaks a leg, they can still visit the center, the physician there will upload the X-ray film to the invited medical institution, and the diagnosis will be conducted remotely. If the center is equipped with a portable X-ray unit, then the patient can be diagnosed at home. Remote medical services promote the intra-group sharing of expertise and ensure that patients living in rural and remote areas have access to the same standards of medical care as those living in urban areas.
iii Online Consultation
Online consultation is the most common type of IPHC provided for first-visit patients with common conditions. Patients can consult physicians or other health professionals at any location about personal medical or psychiatric conditions, or simply seek advice on routine health management, healthy lifestyle, and so on through digital technologies. Online consultation enables patients to receive ongoing care where face-to-face or internet-based diagnoses are not necessary or easily accessible. It is worth pointing out that, while online consultation has much in common with online diagnosis, it lies outside the scope of internet-based diagnosis in the Chinese context. If an online consultation involves diagnosis-making or drug prescriptions, it is indeed classed as an internet-based diagnosis.
Due to space constraints, other IPHC services, such as online health management, electronic medical records management, appointment scheduling, and online payment are not elaborated here.
III Remaining Challenges
Despite considerable progress, the widespread implementation of IPHC remains difficult in practice. Regulatory challenges include restrictions on internet-based diagnosis, physicians practicing at multiple medical institutions, and medical insurance coverage and reimbursement. Technology-related barriers include digital literacy and internet infrastructure, among others. Due to space constraints, the following sections focus on the regulatory challenges.
A Restrictions on Internet-Based Diagnosis
As internet-based diagnosis is a brand-new model of medical service delivery, the NHC has taken a deliberate approach and limited it to “follow-up” diagnoses for “common diseases” and “chronic diseases” in the interests of patient safety and quality of care. Yet, after years of trial implementation, this restriction has raised considerable controversy.
First, the definition and scope of common and chronic diseases is not clear. The AMIDT stipulates that internet-based diagnosis is restricted to “certain” common and chronic diseases, without specifying which diseases fall within that scope. Even though detailed implementation plans of the AMIDT were formulated by provincial health administrations, the wording remained the same. In real practice, the interpretation of this guidance depends largely on the discretion of physicians due to the lack of legal clarity.
Second, it is difficult to verify whether a common or chronic disease was first diagnosed in an offline hospital. According to the AMIDT and other provincial implementation plans, internet hospitals should request to see medical records directly from patients or from other medical institutions with patients’ authorization before diagnosis. Yet, for information security, internet hospitals are less likely to access other institutions’ EMR databases, unless there is a preexisting partnership (e.g., a medical consortium). Patients, in particular the elderly, may neither reserve paper medical records nor understand how to upload their records onto the Internet. In practice, physicians collect patient medical records simply to fulfil regulatory requirements. It is not feasible for them to authenticate patients’ first in-person diagnoses. During the COVID-19 emergency, the NHC lifted the requirement for first in-person diagnoses. Patients with suspected coronavirus symptoms would have an internet-based diagnosis before going to the hospital. This gives rise to the question: Is it still necessary to prohibit internet hospitals from providing a first diagnosis, even just for common or chronic diseases?
After many years of IPHC development, there are plenty of discussions in academia and industry about relaxing the restrictions on the scope of internet-based diagnosis. Arguably, internet hospitals have an obvious limitation: Medical services, such as physical examinations and diagnostic tests, must be conducted in-person in offline hospitals. Therefore, because of quality and safety concerns, strict measures have been taken to regulate the operation of internet hospitals. Prior to the COVID-19 pandemic, internet hospitals just served as a supplement to offline hospitals. Yet the demand for IPHC significantly increased during the health emergency. As such, national and provincial health administrations have issued a number of guidance documents to provide temporary regulatory flexibility, so as to make internet hospitals more widely accessible. Internet hospitals have now become a part of mainstream medical service delivery, and the NHC, in its newly released National Health Informatization Program in the Fourteenth Five-year Plan, intends to set up electronic health files and electronic medical records for every citizen. These provide a good opportunity to conduct further research on the potential and proper trades-offs between convenience of access to health care and safety of service and privacy, while loosening the regulations on internet-based diagnosis.
B Physician Multi-site Practicing
Physician multi-site practicing (PMP) is expected to advance the implementation of IPHC.Footnote 14 PMP is designed as a mechanism to address health professional shortages and improve efficient and equitable allocation of medical resources.Footnote 15 For example, as Haodaifu (“good doctor” in Mandarin) Online (one of the biggest platform-based internet hospitals in China) claims, there are more than 240,000 physicians registered on its platform, more than 70 percent of whom are from tertiary hospitals across the country.Footnote 16 PMP makes it possible for patients living in remote rural areas to receive internet-based diagnoses provided by physicians in big cities, such as Beijing and Shanghai, at home.
PMP needs regulatory clarification. Since the new round of health system reforms in 2009, the Chinese government has issued various guidance documents to encourage physicians to practice at multiple sites. In 2017, the Administrative Measures for the Registration of Practicing Medical Doctors took effect and released limitations on the number and geographic location of medical institutions at which a physician is permitted to practice.Footnote 17 More importantly, the measures simplified the registration procedures for PMP. Approval from the primary practice institution is no longer necessary. Nevertheless, this requirement was once again included in the 2018 Administrative Measures for Internet-based Diagnosis and Treatment. As a result, physicians have to obtain the approval from their primary practice institution before practicing at any internet hospital. Therefore, further clarification is needed regarding the regulation of PMP.
To a lesser extent, even though prior approval would not be necessary for multi-practicing online, it does not mean that the primary practice institution has no de facto discretion when it comes to PMP. In China, most physicians are hired by medical institutions (in most instances their primary practice institution) and are, thus, subject to the personnel management of the institution. Physicians are the most valuable medical resources and the core competence of any medical institution. Arguably, PMP would have a considerable impact on the operation of the primary practice institution. Also, given the high workload in public medical institutions and especially tertiary hospitals, some institutions may take administrative measures to restrict de facto PMP, except for in their affiliated online or offline institutions.
There are also concerns over the affordability and quality of care regarding PMP. On the one hand, prices for medical services provided by public medical institutions, whether online or offline, are capped by governments, while those provided by non-public medical institutions (e.g., platform-based internet hospitals) are self-determined. In addition, as will be discussed further in the next section, medical services provided by non-public medical institutions are generally not covered by the country’s mandatory basic medical insurance (BMI) schemes, unless these institutions choose to negotiate prices and sign contracts with local health insurance bureaus (i.e., insurers).Footnote 18 Consequently, the service fees charged by non-public medical institutions are higher than public ones and patients have to pay all the service fees out-of-pocket, unless the patient has extra commercial insurance to cover all or parts of the expenses. For instance, for the same specialist, the outpatient service fee charged by the aforementioned Haodaifu Online could be ten times higher than that of the tertiary hospital. On the other hand, physicians are better paid under this circumstance. Financial incentives could motivate them to allocate more (free) time to platform-based internet hospitals, and waiting times for hospital admission would, thus, be significantly reduced. However, there is a dilemma for patients: PMP at platform-based internet hospitals makes medical services provided by specialists more accessible, yet less affordable. In other words, patients need to pay more money in exchange for a shorter waiting time for specialist medical services. PMP may also have a negative impact on the quality of care provided by the specialist’s primary practice institution. Therefore, national and provincial health administrations have made some principal guidelines on PMP, requiring physicians to give priority to the work at their primary practice institution. It would thus be important for policy makers to consider complementary measures to encourage as well as to regulate PMP, so as to further improve the accessibility, affordability and quality of health care.
C Affordability of IPHC
Affordability is one of the key determinants of IPHC. The NHSA has, therefore, issued a series of policies to make IPHC more affordable to patients. In 2019, the NHSA announced for the first time that all eligible “internet plus” medical services would gradually be covered by medical insurance in the Guiding Opinions on Improving the “Internet Plus” Medical Service Price and Medical Insurance Coverage Policy.Footnote 19 This document authorizes provincial health insurance bureaus to set prices for internet-based diagnoses and other medical services provided by public medical institutions, while non-public medical institutions are allowed to set their own service prices. Nevertheless, prior to the COVID-19 pandemic, internet-based medical services had not started to be covered by BMI schemes.
In response to the COVID-19 outbreak, the NHSA and NHC issued their Guiding Opinions on Promoting “Internet Plus” Medical Insurance Service during the Prevention and Control of COVID-19, expanding the BMI coverage to make internet-based diagnosis and other medical services more affordable.Footnote 20 The pricing policy remains unchanged in this guidance document. BMI programs would cover and reimburse internet-based diagnoses for common and chronic diseases provided by designated public medical institutions that voluntarily signed a supplementary contract with local health insurance bureaus. Internet-based diagnoses provided by designated non-public medical institutions would also be reimbursed, but at the same rate as public medical institutions if non-public institutions choose to provide such services. Multiple provinces and municipalities have also taken actions to temporarily broaden provincial BMI schemes to cover internet-based diagnoses, as well as expand the types of internet hospitals which may provide such services.
Several months later, the NHSA issued another document, Guiding Opinions on Actively Promoting Medical Insurance Coverage of “Internet Plus” Medical Service, establishing concrete measures to promote the reimbursement and coverage of internet-based medical services.Footnote 21 The new Guiding Opinions make clear that a voluntary supplementary contract between designated medical institutions and local health insurance bureaus is a prerequisite for BMI coverage. Payment parity is granted, which means that internet-based diagnoses will be reimbursed at the same rate as the equivalent in-person services provided by public medical institutions offline. However, this document does not require service parity. Provincial health insurance bureaus are authorized to determine the coverage of services in their own insurance plans. Research suggests that twenty-one Chinese provinces have so far expanded their provincial BMI coverage of internet-based diagnoses, while the scope of coverage varies from province to province.Footnote 22 In addition, as just explained, non-public medical institutions could set their own pricing for medical services, no matter whether they are provided online or offline. Such services will not be covered nor reimbursed by BMI schemes, unless these institutions choose to negotiate prices and sign contracts with local health insurance bureaus.Footnote 23
To sum up, affordability was, is, and may still be a major barrier for the utilization of IPHC. Although the NHSA has issued a number of polices to expand coverage, most of them only provide principal guidelines, without an integrated regulatory framework for “internet plus” health insurance coverage and reimbursement.
IV Conclusion: The Way Forward
IPHC has proven to be critical and full of potential for strengthening the Chinese health system, transforming health care services, and improving equitable, affordable, and universal access to health. The Chinese government has taken a variety of measures to accelerate the utilization of IPHC before, during, and after COVID-19, such as the establishment and revision of regulations, the removal of restrictions, and adjustments to reimbursement mechanisms. However, gaps remain in the legal and regulatory framework for governing the use of IPHC. Many of the reimbursement mechanisms have been established as exceptions rather than permanent changes. Also, most IPHC-related regulations are still in trial the phases of implementation. Therefore, it is necessary to develop a clear legal and regulatory framework for supporting the development and sustained use of IPHC, and for eventually developing an “internet plus” health ecosystem in the post-COVID-19 era. Additional research on the potential trades-offs in loosening the regulations on internet-based diagnoses, as well as PMP, is needed. In addition, the use of digital technologies for health helps to improve geographic access to health, yet it may exacerbate other inequalities due to digital literacy. For example, the elderly living alone face greater challenges when it comes to using digital technologies to access internet hospitals. Further research should pay particular attention to the special needs of vulnerable groups and focus on how to improve their digital literacy and access to the Internet. Also, additional studies on how to strike the balance between data sharing and privacy protection are much needed.














 Open access
Open access