


- ACC
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- mTOR
mammalian target-of-rapamycin
Animal cells take up fuel molecules such as glucose or fatty acids, and oxidise them to CO2 via the process of catabolism. Much of the energy released during this process is used to convert ADP to ATP, which can be likened to the chemicals in a rechargeable battery. Extending this analogy, catabolism charges up the battery by converting ADP to ATP, whereas most other cellular activities (e.g. growth, division, secretion and movement) require energy and are driven by the conversion of ATP back to ADP, thus draining the battery. Just as machines that utilise rechargeable batteries (such as laptop computers or electric cars) require systems to monitor the state of the battery, cells require systems to monitor their ATP:ADP ratio and match the rates of uptake and consumption of carbon nutrients to the rate of ATP utilisation. The topic of this review is the AMP-activated protein kinase (AMPK), which is the major system responsible for achieving this task in eukaryotes. Researchers who wish to understand disorders of energy balance, such as obesity and type-2 diabetes, have been particularly interested in the system.
Protein kinases, including AMPK, are signalling enzymes that modify the function of target proteins by transferring phosphate groups from ATP to side chains of specific amino acids, usually serine or, less frequently, threonine or tyrosine. This modification, which is reversed by a different reaction catalysed by protein phosphatases, often triggers a marked change in the function of the target protein. For example, if the target is a metabolic enzyme it can switch enzyme activity on or off, or change the response to other regulators. In other cases, phosphorylation targets proteins for degradation, changes their association with other proteins, or affects their targeting to particular subcellular compartments. The importance of protein kinases was emphasised when sequencing of the human genome revealed that it encoded over 500 protein kinase catalytic subunits, accounting for nearly 2% of all genes(Reference Manning, Whyte and Martinez1). This great diversity reflects the likelihood that protein phosphorylation is the most important mechanism by which cell function is modulated in response to changes in local conditions, and to molecules like hormones and cytokines that carry messages about conditions in other locations.
The AMPK was first defined in the late 1980s by the author when he recognised that protein kinases previously found to phosphorylate and inactivate the key regulatory enzymes of fatty acid and cholesterol synthesis, i.e. acetyl-CoA carboxylase (ACC) and 3-hydroxy-3-methylglutaryl-CoA reductase, were identical(Reference Carling, Zammit and Hardie2). We named it AMPK after the nucleotide 5′-AMP, which causes its activation(Reference Hardie, Carling and Sim3). Why should cells express a protein kinase activated by AMP? A high ATP:ADP ratio, which is maintained by catabolism in cells that have adequate supplies of glucose and oxygen, drives the reversible reaction catalysed by adenylate kinase (ATP+AMP↔2ADP) from left to right, keeping the AMP concentration low (up to 100-fold lower than ATP). However, any metabolic stress that causes a drop in the ATP: ADP ratio will cause some displacement of the adenylate kinase reaction to the left, producing AMP (Fig. 1). A rise in AMP is therefore a signal that the energy status of the cell has been compromised. Since deprivation for glucose or oxygen are two of the metabolic stresses that can cause an increase in AMP, the AMPK system acts as a nutrient sensor and is a key player in cellular sensing mechanisms for both glucose and oxygen.
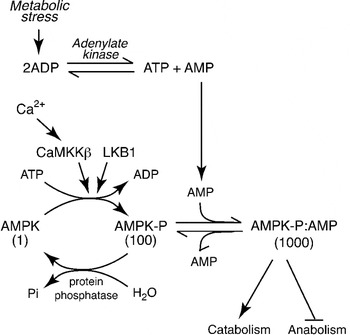
Fig. 1. Regulation of the AMP-activated protein kinase (AMPK) system. Numbers below the three forms of AMPK indicate their relative kinase activity. The upstream kinase (LKB1) continually converts AMPK to its phosphorylated form, increasing the activity 100-fold. However, in the absence of AMP it is rapidly converted back to the dephosphorylated, inactive form by protein phosphatases. Binding of AMP causes a conformational change that increases the activity a further 10-fold via an allosteric effect, and also prevents dephosphorylation. The activating signal, AMP, rises during metabolic stress because a rise in ADP causes displacement of the adenylate kinase reaction towards AMP. The system can also be activated by a rise in intracellular Ca2+, due to phosphorylation catalysed by the Ca2+-activated kinase calmodulin-dependent kinase kinase-β (CaMKKβ).
Regulation, structure and evolution of AMP-activated protein kinase
Addition of AMP causes almost instant activation of AMPK by up to 10-fold(Reference Carling, Zammit and Hardie2, Reference Suter, Riek and Tuerk4). Since this is caused by binding of AMP at a site distinct from the active site, it is referred to as allosteric activation. However, it is not the whole story: the author discovered that phosphorylation of AMPK at a specific site within the kinase domain (Thr-172), catalysed by distinct upstream protein kinases, causes >100-fold activation of AMPK(Reference Hawley, Davison and Woods5) (Fig. 1). This multiplies with the allosteric effect to give up to 1000-fold activation overall. Phospho-specific antibodies recognising AMPK phosphorylated at Thr-172, and antibodies recognising the downstream target ACC phosphorylated at the AMPK site (Ser-79) are now widely used as biomarkers to monitor AMPK activation. We and others identified the major upstream kinase phosphorylating Thr-172 to be a complex containing the protein kinase LKB1(Reference Hawley, Boudeau and Reid6, Reference Woods, Johnstone and Dickerson7). This was an exciting discovery, because LKB1 had been previously identified as a tumour suppressor, i.e. a gene product that inhibits the development of cancer. It now seems clear that AMPK mediates many of the tumour suppressor effects of LKB1, although discussion of this is beyond the scope of this review. The LKB1 complex appears to phosphorylate Thr-172 continuously under basal conditions, although the phosphate is normally immediately removed by protein phosphatases. However, the binding of AMP to AMPK causes a structural change that prevents dephosphorylation of Thr-172, thus triggering a net switch to the phosphorylated form, causing an activation that is further amplified by the allosteric mechanism. Thr-172 can also be phosphorylated by the Ca2+-activated kinase calmodulin-dependent kinase kinase-β, which can trigger the activation of AMPK in response to agents that increase intracellular Ca2+ (Fig. 1), even in the absence of a rise in AMP(Reference Hawley, Pan and Mustard8, Reference Woods, Dickerson and Heath9).
AMPK occurs universally as heterotrimeric complexes, i.e. complexes formed from three non-identical protein subunits, these being the catalytic α subunit (which contains Thr-172) and the regulatory β and γ subunits(Reference Hardie10). In human subjects each subunit is encoded by multiple genes, giving rise to up to twelve heterotrimeric combinations whose expression varies between cell types. The β subunits contain carbohydrate-binding modules whose regulatory function will be discussed later, whereas the γ subunits contain two sites that bind the regulatory nucleotide, AMP(Reference Scott, Hawley and Green11, Reference Xiao, Heath and Saiu12). Why there should be two sites is not yet clear, but one explanation is that one is required for the allosteric effect of AMP and the other for the effect on dephosphorylation. Both sites also bind ATP in competition with AMP, but this fails to trigger the activating effects. Thus, AMPK is activated by an increase in the cellular AMP:ATP ratio, a signal that the energy status of the cell is compromised.
Genes encoding the α, β and γ subunits of AMPK are readily found in the genomes of all eukaryotes, ranging from unicellular organisms such as protozoa (e.g. Giardia lamblia) and fungi (e.g. budding yeast, Saccharomyces cerevisae), to multicellular organisms such as nematodes, insects, plants and mammals. In lower eukaryotes, the function of the AMPK relatives (orthologues) has been studied by genetics, and the results give interesting clues as to the probable function of AMPK during early evolution. Budding yeast lacking the complex are viable if always kept in a medium containing high glucose, but cannot mount any of the normal responses to glucose removal, showing that it is involved in nutrient sensing and the response to starvation(Reference Hardie, Carling and Carlson13). In the nematode worm Caenorhabditis elegans, the AMPK orthologue is involved in response to dietary energy restriction. This organism was one of the model systems where the effect of dietary energy restriction to enhance longevity, which is now thought to occur also in mammals, was first defined. Restricting the diet during early development delays sexual maturation of the worms but also considerably extends their lifespan, and the AMPK orthologue is required for this response(Reference Apfeld, O'Connor and McDonagh14, Reference Narbonne and Roy15). In the plant Physcomitrella patens (a moss), the AMPK orthologue is not required for growth in continuous light, but is required for growth in alternate light–dark cycles(Reference Thelander, Olsson and Ronne16). Since darkness for a plant is the equivalent of a period of starvation or fasting, this reinforces the view that the ancestral role of the AMPK system was in response to starvation for carbon nutrients.
Activation of mammalian AMP-activated protein kinase by metabolic stresses
If mammalian cells are deprived of glucose in culture, AMPK is rapidly activated (as in yeast) due to a reduced rate of glucose catabolism and a consequent increase in the AMP:ATP ratio(Reference Salt, Johnson and Ashcroft17). Most mammalian cells express GLUT1 or GLUT4, and the first step in subsequent glucose metabolism is catalysed by hexokinase. GLUT1, GLUT4 and hexokinase all exhibit high Km values for glucose, and are thus saturated at normal plasma glucose concentrations. In these cells, the activation of AMPK therefore only occurs at very low glucose concentrations that may not be physiologically relevant. However, specialised ‘glucose-sensing’ cells, such as the β cells of the pancreas and specific neurones in the hypothalamus, express high Km transporters and glucose phosphorylating enzymes, i.e. GLUT2 and glucokinase, and fluctuations in glucose within the normal physiological range alter the rate of glucose metabolism and consequently modulate the AMPK activity. In these cells, the activation of AMPK appears to play important roles in the physiological responses to hypoglycaemia, such as reduced secretion of insulin and increased secretion of glucagon and adrenaline by the pancreas and adrenal medulla(Reference Salt, Johnson and Ashcroft17–Reference McCrimmon, Shaw and Fan20).
Deprivation of oxygen (hypoxia) is another stress that can limit catabolism, thus causing increases in AMP:ATP and AMPK activation. This occurs, for example, in cardiac muscle in response to ischaemia, an interruption in the blood supply(Reference Kudo, Barr and Barr21–Reference Russell, Li and Coven23). Similar to what has just been described for hypoglycaemia, in most cells the oxygen tension has to drop to pathologically low levels (such as occurs during ischaemia) before AMPK is activated. However, just as there are specialised glucose-sensing cells, there are also specialised oxygen-sensing cells, such as the Type 1 cells in the carotid body, in which AMPK is activated by more physiological levels of hypoxia. Type 1 cells release neurotransmitters onto afferent nerves, signalling to the brain to regulate breathing in response to the oxygen content of the blood reaching it via the carotid artery. Other oxygen-sensing cells include the smooth muscle cells lining pulmonary arteries, which (opposite to what happens in most arteries) contract in response to hypoxia, in order to divert blood flow to more oxygenated regions of the lung. In both cell types, mitochondrial catabolism appears to be particularly sensitive to decreasing oxygen tension so that AMPK is activated, triggering changes in Ca2+ movement that promote either neurotransmitter release (Type 1 cells) or contraction (pulmonary smooth muscle)(Reference Evans, Mustard and Wyatt24–Reference Wyatt, Pearson and Kumar26).
The stresses discussed thus far activate AMPK by inhibiting ATP synthesis. A metabolic stress that activates AMPK by accelerating ATP consumption, and which occurs under normal physiological conditions, is muscle contraction. With Will Winder, the author found that AMPK is activated in rodent skeletal muscle during treadmill exercise(Reference Winder and Hardie27), and this has been confirmed many times using biopsies of human muscle. In a later section, I will focus on the metabolic effects that occur downstream of AMPK in contracting muscle.
Regulation of AMP-activated protein kinase by adipokines and other cytokines
As discussed earlier, the ancestral role of AMPK in unicellular eukaryotes appears to have been in response to starvation for a carbon source. In mammals, plasma glucose is maintained within tight limits by hormonal homoeostasis, and so glucose deprivation is less of an issue. Nevertheless, AMPK is still ubiquitously expressed. It appears that during the evolution of multicellular organisms, adaptations occurred to allow other types of input to interact with the system. This is illustrated by the effects of the adipokines, cytokine-like substances released by adipocytes, of which leptin is the classical example. Leptin activates AMPK in skeletal muscle, accounting for its ability to stimulate fatty acid oxidation (see the next section) and hence energy expenditure(Reference Minokoshi, Kim and Peroni28). The more well-known role of leptin is to repress appetite (and hence energy intake) via effects on the hypothalamus, and Kahn and co-workers have suggested that this is mediated by inhibition of AMPK(Reference Minokoshi, Alquier and Furukawa29). Results with knock-out mice that lack AMPK in hypothalamic neurones have not fully supported this model, but do support the idea that AMPK is involved in glucose sensing in the hypothalamus(Reference Claret, Smith and Batterham19). There is also widespread agreement that treatments which activate AMPK in the hypothalamus, such as infusion with the nucleoside 5-aminoimidazole-4-carboxamide riboside, treatment with the gut hormone ghrelin or cannabinoids, or hypoglycaemia, all increase food intake in rodents(Reference Andersson, Filipsson and Abbott30–Reference McCrimmon, Fan and Ding32).
The other well-known adipokine is adiponectin, whose plasma concentration (in contrast to leptin) is paradoxically reduced in obese or insulin-resistant individuals. Adiponectin, acting via the AdipoRI receptor(Reference Yamauchi, Kamon and Ito33), activates AMPK in the liver and other tissues, and this appears to be responsible for many of its effects, including suppression of gluconeogenesis in the liver(Reference Yamauchi, Kamon and Minokoshi34) and enhancement of food intake via effects on the hypothalamus(Reference Kubota, Yano and Kubota35). Finally, a number of cytokines, hormones and nutrients have been reported to modulate AMPK activity, accounting for diverse effects in different cell types, including IL-6(Reference Carey, Steinberg and Macaulay36), ciliary neurotrophic factor(Reference Steinberg, Watt and Fam37, Reference Watt, Dzamko and Thomas38), macrophage inhibitory factor(Reference Miller, Li and Leng39) and lipoic acid(Reference Kim, Park and Namkoong40). In most of these cases, the exact mechanisms by which these agents activate AMPK, whether through changes in AMP:ATP or some other mechanism, remain unclear.
Metabolic effects of AMP-activated protein kinase activation during muscle contraction
The metabolic consequences of AMPK activation have been particularly well studied in skeletal muscle. AMPK is responsible not only for acute responses to exercise, occurring within seconds or minutes, but also for longer-term adaptations to repeated exercise caused by changes in gene expression. AMPK is mainly activated during prolonged exercise and may not be involved in responses to short bouts of resistance exercise, such as weight-lifting. In this case, much of the ATP is generated by the conversion of phosphocreatine to ATP, so that there is little change in the content of ATP, ADP or AMP. As phosphocreatine becomes depleted during an event such as a sprint, glycogen breakdown and anaerobic glycolysis then become important for the generation of ATP. However, once again these effects do not require AMPK, because the key regulatory enzymes involved (phosphorylase and phosphofructokinase) directly bind AMP and are activated by increases in the AMP:ATP ratio independent of AMPK. AMPK does become crucial in the switch from anaerobic to aerobic metabolism that is required for endurance exercise, for example, in running events of more than 200 m. During prolonged exercise, the use of aerobic metabolism and blood-borne fuels such as glucose and fatty acids becomes increasingly important, and this is where AMPK plays a key role. Intriguingly, one of the roles of the AMPK orthologue in budding yeast is to trigger the switch from fermentation (anaerobic metabolism) to the more efficient oxidative metabolism when the glucose in the medium starts to become depleted(Reference Hardie, Carling and Carlson13).
The acute increase in glucose uptake in skeletal muscle in response to contraction is caused by a translocation of the GLUT4 from intracellular storage vesicles to the plasma membrane. The first evidence that this might be mediated by AMPK came from experiments by Winder and the author using the nucleoside, 5-aminoimidazole-4-carboxamide riboside. This is taken up by cells and converted to the nucleotide 5-aminoimidazole-4-carboxamide riboside monophosphate, which mimics the activating effects of AMP on the AMPK system. When rat skeletal muscle was perfused with 5-aminoimidazole-4-carboxamide riboside, AMPK was activated and this was associated with increased glucose uptake(Reference Merrill, Kurth and Hardie41). Subsequent studies with GM mice, in which AMPK activation during contraction was rendered defective by various means, confirmed that AMPK was involved in this effect(Reference Mu, Brozinick and Valladares42, Reference Sakamoto, McCarthy and Smith43). There may be multiple mechanisms by which AMPK promotes GLUT4 translocation to the plasma membrane, but one appears to be the phosphorylation of a protein called TBC1D1 (TRE2/BUB2/CDC16 domain family member 1), which enhances GLUT4 translocation by promoting the conversion of small guanine nucleotide-binding proteins called Rabs from their inactive (Rab:GDP) to their active (Rab:GTP) forms(Reference Chen, Murphy and Toth44, Reference Pehmoller, Treebak and Birk45). This is similar to the mechanism by which insulin stimulates glucose uptake in resting muscle, when the insulin-activated protein kinase B phosphorylates TBC1D1 or other members of the same family. In insulin-resistant individuals, insulin-dependent activation of protein kinase B is defective, but the mechanism by which contraction stimulates glucose uptake via AMPK remains unaffected(Reference Musi, Fujii and Hirshman46). This is one reason why regular exercise is particularly beneficial in insulin-resistant subjects.
Another catabolic pathway activated during low-intensity exercise is fatty acid oxidation. Muscle expresses the ACC2 isoform of ACC which, like the ACC1 isoform expressed in most other cells, is phosphorylated and inactivated by AMPK(Reference Winder, Wilson and Hardie47). ACC2 is associated with mitochondria and produces malonyl-CoA, an inhibitor of the enzyme carnitine-palmitoyl transferase-1 located on the outer mitochondrial membrane. Carnitine-palmitoyl transferase-1 is required for the uptake of fatty acids into mitochondria, and is inhibited in resting muscle by the malonyl-CoA produced by ACC2(Reference McGarry and Brown48). Inhibition of ACC2 by AMPK during contraction therefore promotes fatty acid uptake into mitochondria and hence oxidation(Reference Merrill, Kurth and Hardie41). Interestingly, excessive storage of fatty acids as TAG in muscle, as in other tissues, is associated with insulin resistance. Stimulation of fat oxidation by AMPK is therefore another reason why exercise is beneficial in insulin-resistant subjects.
As well as these acute effects on muscle metabolism, AMPK is also responsible for some of the longer-term adaptations induced by repeated endurance exercise training. This includes increased expression of GLUT4(Reference Zheng, MacLean and Pohnert49), which may be mediated in part by phosphorylation of a histone deacetylase that modifies the chromatin structure at the GLUT4 promoter(Reference McGee, van Denderen and Howlett50). AMPK activation also up-regulates mitochondrial biogenesis by up-regulating the transcriptional co-activator PPARγ co-activator-1α, which promotes the expression of mitochondrial genes encoded in both nuclear and mitochondrial DNA. AMPK has been reported to directly phosphorylate PPARγ co-activator-1α(Reference Jager, Handschin and St-Pierre51), but the mechanism may also involve deacetylation of PPARγ co-activator-1α by Sirtuin-1, which is activated downstream of AMPK(Reference Canto, Gerhart-Hines and Feige52).
These longer-term adaptations to endurance exercise would ensure that, as exercise is repeated regularly, it becomes easier because glucose is taken up more rapidly and mitochondrial ATP production is faster. Because both glucose uptake and mitochondrial function tend to be reduced in individuals at risk of developing type-2 diabetes(Reference Lowell and Shulman53), AMPK can also help to explain the ability of regular exercise to provide protection against the development of diabetes.
Muscle contraction causes very rapid ATP consumption and it therefore makes sense that other ATP-requiring processes, such as biosynthesis, should be switched off. Another target for AMPK in skeletal muscle is glycogen synthase, the enzyme catalysing the last step in glycogen synthesis that also controls its rate(Reference Carling and Hardie54, Reference Jorgensen, Nielsen and Birk55). AMPK phosphorylates glycogen synthase at site 2, reducing its activity(Reference Carling and Hardie54). This may help to ensure that when glucose uptake increases during contraction, the increased flux of glucose into the cell is diverted towards catabolic breakdown of glucose rather than glycogen synthesis. Paradoxically, however, immediately after exercise glycogen synthase is found to be in a highly dephosphorylated and active state(Reference Danforth56). The β subunits of AMPK contain carbohydrate-binding modules that cause the complex to associate with glycogen particles in intact cells and in vivo (Reference Hudson, Pan and James57–Reference Bendayan, Londono and Kemp59). While the role of the carbohydrate-binding module may in part be simply to localise AMPK in close proximity to its target, glycogen synthase, the author has recently obtained evidence that glycogen also inhibits AMPK(Reference McBride, Ghilagaber and Nikolaev60). Synthetic oligosaccharides that mimic the non-reducing ends of glycogen at the surface of the glycogen particle inhibit the kinase activity(Reference McBride, Ghilagaber and Nikolaev60), and recent unpublished evidence shows that the inhibition reaches a maximum using a chain of six glucose units lacking a non-reducing end (C Chevtzoff and DG Hardie, unpublished results). Our current hypothesis is that, in a full-sized glycogen particle that has up to 2000 non-reducing ends packed around the surface of a glycogen particle that is only 20–30 nm in diameter(Reference Melendez-Hevia, Waddell and Shelton61), the non-reducing ends would be so tightly packed that they would be unable to inhibit AMPK, although it may still bind to the particle via the carbohydrate-binding module. In a full-sized glycogen particle AMPK would thus phosphorylate glycogen synthase, preventing it from further extending the outer chains of glycogen. However, as glycogen becomes degraded during exercise the outer branches would be removed and the structure would become more open. AMPK would now become inhibited due to the greater accessibility of the non-reducing ends, and would no longer phosphorylate glycogen synthase, which would instead be dephosphorylated by protein phosphatases bound to the glycogen particle(Reference Aschenbach, Suzuki and Breeden62). This model provides a potential explanation for the paradoxical observation that, although AMPK is activated during exercise, its substrate glycogen synthase is found to be dephosphorylated immediately following exercise(Reference Danforth56, Reference Aschenbach, Suzuki and Breeden62). This mechanism would ensure that, if glycogen became significantly depleted, it would be rapidly replenished as soon as exercise ceased. If this model is correct, AMPK not only monitors the short-term availability of energy in the form of ATP and ADP, but also the medium term reserves in the form of glycogen. It also means that AMPK is involved in the rapid glucose uptake that occurs following exercise to replenish glycogen, another of the beneficial outcomes of exercise in individuals who are insulin resistant or hyperglycaemic.
Another biosynthetic pathway that is switched off, at least temporarily, during exercise is translation of mRNA into protein. An important signalling pathway that stimulates translation has as its central component the mammalian target-of-rapamycin (mTOR). mTOR is a protein kinase that is stimulated by insulin and by amino acids, and it promotes protein translation by regulating its initiation(Reference Tee and Blenis63). AMPK activation, on the other hand, inhibits mTOR by phosphorylating multiple upstream regulators(Reference Inoki, Zhu and Guan64, Reference Gwinn, Shackelford and Egan65), and thus inhibits protein synthesis. There is evidence that this mechanism does partly account for a temporary cessation of translation in muscle during contraction(Reference Bolster, Crozier and Kimball66, Reference Rose, Bisiani and Vistisen67). Interestingly, mTOR is also activated during resistance exercise training(Reference Baar and Esser68) as well as by insulin and by feeding amino acids, and this may account for the protein synthesis and hence muscle hypertrophy induced by all of these inputs. Since AMPK opposes mTOR activation, this may help to explain why endurance exercise training does not cause muscle hypertrophy, and why endurance athletes have less bulky muscles compared to weight-lifters or sprinters.
Regulation of AMP-activated protein kinase by drugs and ‘nutraceuticals’
In the previous section, I stressed the numerous metabolic changes caused by AMPK activation in muscle, which are particularly beneficial in individuals at risk of developing insulin resistance and/or type-2 diabetes. AMPK activation in the liver, which occurs in response to adiponectin, is also beneficial by repressing production of glucose by gluconeogenesis(Reference Yamauchi, Kamon and Minokoshi34, Reference Lochhead, Salt and Walker69), which is at least partly responsible for the tendency of insulin-resistant individuals to hyperglycaemia. AMPK activation in the liver also promotes fat oxidation(Reference Yamauchi, Kamon and Minokoshi34), thus reducing TAG accumulation, a metabolic state that is strongly associated with insulin resistance. It is therefore not surprising that AMPK has become an exciting target for new drugs aimed at the treatment of type-2 diabetes(Reference Hardie70). In fact, the biguanide metformin, which was developed from an ancient herbal remedy and is now the first choice drug for treatment of type-2 diabetes, activates AMPK(Reference Zhou, Myers and Li71), and there is good evidence from rodents that this accounts for its ability to lower plasma glucose(Reference Shaw, Lamia and Vasquez72). The author has recently shown that metformin activates AMPK indirectly by causing a mild inhibition of complex I of the respiratory chain, thus increasing cellular AMP:ATP(Reference Hawley, Ross and Chevtzoff73). Despite this rather crude mechanism of action, metformin is a safe drug that is now prescribed to >100 million patients worldwide. A number of other xenobiotics or ‘nutraceuticals’ derived from foods and beverages, or from traditional herbal medicines, also activate AMPK and have been claimed to have beneficial effects in obesity, type-2 diabetes and even cancer. These include resveratrol, present in small quantities in red wine, quercetin present in many fruit and vegetables and berberine, a yellow dye derived from plants of the genus Berberis used in traditional Chinese medicine. We have recently reported that all of these activate AMPK due to their ability to cause inhibition of mitochondrial ATP production(Reference Hawley, Ross and Chevtzoff73). They are all secondary metabolites of plants, and an interesting speculation is that they may be produced by the plant to prevent or discourage grazing by animals, or infection by micro-organisms. In support of this, the natural product galegine (from which metformin is derived and which itself a potent AMPK activator(Reference Hawley, Ross and Chevtzoff73)), is produced by the plant Galega officinalis, which also goes by the name of Goat's Rue and is classed as a noxious weed in USA because it is poisonous to herbivores. Also, resveratrol is produced by grapes in response to fungal infection, which is why its concentration is often higher in organic wines where the grapes have not been treated with fungicides.
Conclusions
AMPK is an ancient system that arose during early eukaryotic evolution, where its role may have been in response to starvation for a carbon source. In mammals, by monitoring the rate of catabolism, it can act in specialised cells as a sensor of either glucose or oxygen. In skeletal muscle it is activated by contraction and appears to be responsible for many of the acute metabolic changes and the longer-term adaptations induced by exercise. Through its regulation by adipokines and other signal molecules, AMPK is also a regulator of energy balance at the whole body level, and drugs that are used to treat disorders of energy balance, such as metformin, now appear to work by activating AMPK.
Acknowledgements
The author declares no conflicts of interest. Studies in the author's laboratory were supported by Programme Grants (080982; 081195) from the Wellcome Trust, and by the companies (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck KGaA and Pfizer) that support the Division of Signal Transduction Therapy at the University of Dundee.

