


Dietary and other lifestyle factors substantially influence colorectal cancer (CRC) risk(Reference Doll and Peto1) and they seem to be responsible for the large differences in CRC incidence around the world(Reference Parkin2). Since Burkitt observed in 1969 an inverse correlation between fibre intake and CRC(Reference Burkitt3), a significant number of studies have supported this protective role(Reference Dahm, Keogh and Spencer4–Reference Howe, Benito and Castelleto6). Several mechanisms have been suggested to explain the presumed preventive effect. Thus, reduction of toxin exposure to the colonic mucosa through dilution of luminal contents and/or faster transit time; bile acid adsorption; and the capacity to increase the generation of SCFA in the gut have all been proposed as potential mechanisms(Reference Corfe, Williams and Bury7). SCFA are produced through the fermentation of fibre by colonic bacteria. The main SCFA generated are acetate, propionate and butyrate. The latter is the major energy source for healthy colonocytes and it is thought to be implicated in the presumable protective effect as it has been shown to promote differentiation, cell-cycle arrest and apoptosis in transformed colonocytes(Reference Wong, de Souza and Kendall8). In an in vivo model of experimentally induced CRC, a higher faecal butyrate concentration was significantly associated with a lower number of tumours developed(Reference McIntyre, Gibson and Young9).
While there are plenty of convincing data, not all studies have been able to prove the chemo-preventive effect of fibre on CRC and in fact all randomised controlled trials have failed to do so(Reference Park, Hunter and Spiegelman10–Reference Bonithon-Kopp, Kronborg and Giacosa12). Relatively short interventions, follow-up times and type of fibre consumed could underlie the lack of significant effect in these trials(Reference Martinez, Marshall and Giovannucci13). Regarding the type of fibre, several studies have supported the protective role of Plantago ovata husk (PO), a partially fermentable, 70 % soluble fibre. Thus, in a paired case–control study, and after adjusting for diet, smoking, alcohol consumption, family history of cancer and socio-demographic factors, consumption of PO, physical exercise and aspirin were all significantly associated with a reduction in CRC risk(Reference Juarranz, Calle-Puron and Gonzalez-Navarro14). A recent comparative ecological study showed an inverse correlation between PO consumption and mortality from CRC that reached statistical significance for the top quintile of PO consumption in the adjusted analysis(Reference Lopez, Villanueva and Martinez-Hernandez15).
In the present study, we proposed to assess the capacity of the fermentation products of PO to induce apoptosis, and to study the mechanisms implicated in this effect. In order to maximally resemble a physiological environment, PO was incubated with human stool in anaerobic conditions and the totality of the resulting products was used to treat colonocytes and assess the effects. Immediately after fermentation, SCFA were measured in the fermentation samples. As reported before, PO fermentation with human faecal flora results in up to three times increase in SCFA production(Reference Fernandez-Banares, Hinojosa and Sanchez-Lombrana16), and we hypothesised that PO's chemo-preventive effects could be due to this significant increase in the generation of SCFA.
Materials and methods
Plantago ovata fermentation and SCFA quantification
PO fermentation and quantification were based on a modified protocol by Clausen et al. (Reference Clausen, Bonnen and Mortensen17). Thus, fresh stool samples were obtained from healthy human volunteers and homogenised with 150 mm-NaHCO3 buffer in a 1:5 ratio. Stool was incubated with Plantaben® (Rottapharm, SL, Spain) and vortexed. Plantaben® contains PO (70 % soluble fibre) and excipients (tartaric acid and sodium bicarbonate). Faecal homogenates had a final concentration of 20 mg/ml of PO as used in previous studies(Reference Nordgaard, Hove and Clausen18). The concentration used is based on average concentrations found in stool after taking sachets of 5 g of Plantaben® containing a total of 7–10·5 g of PO (M Eastwood, unpublished results). Anaerobic fermentation was achieved by the exchange of air for nitrogen in the sealed tubes. Tubes were then incubated at 37°C in a shaking water bath for 72 h. Control tubes without PO powder addition were fermented in the same way. All tubes were stored at − 80°C after fermentation.
For SCFA quantification, fermented samples were acidified with H2SO4 and centrifuged to take apart solid material. The supernatant was then recovered and SCFA were extracted by adding diethyl ether, vortexing for 1 min and allowing the phases to separate. The diethyl ether phase containing the fatty acids was recovered. Quantification was performed using a JEOL GCmate™ II GC/MS double-focusing mass spectrometer (JEOL Limited Tokyo, Japan). As an internal standard, 2-methylvaleric acid was used. A standard curve was performed using six samples with known concentrations of acetic, propionic, butyric, isobutyric, isovaleric, valeric and caproic acids and extracted with diethyl ether. All samples were analysed twice each for acetate, butyrate and propionate concentrations. Table 1 shows the concentrations of SCFA in stool with no PO added and after 72 h of PO stool fermentation. The averages of butyrate measurements were used to determine cell treatment concentrations.
Table 1 Generation of SCFA after stool fermentation with Plantago ovata husk (PO)*
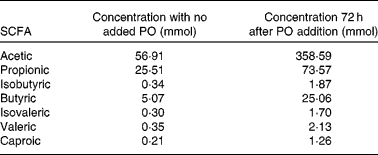
* Data shows SCFA concentrations after fermentation for 72 h at 37°C in anaerobic conditions.
The study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects were approved by the Hospital Universitari Germans Trias i Pujol ethics committee. Written informed consent was obtained from all subjects.
Cell cultures and supplements
To study the effects of PO, we used five human colon adenocarcinoma cell lines with different molecular phenotypes, such as wild-type p53 (HCT116, LoVo) v. mutated p53 (Caco-2, HT-29, SW480); microsatellite stable (Caco-2, HT-29, SW480) v. microsatellite unstable (HCT116, LoVo); wild-type β-catenin (LoVo, SW480, HT-29) v. mutated β-catenin (HCT116, Caco-2); primary tumour, non-metastatic (HCT 116, Caco-2, HT-29, SW480) v. metastatic (LoVo). Table 2 shows some of the main features of the cell lines studied. Doubling times in CRC cells ranged from 20 h for HCT116 to 62 h for Caco-2. Cell lines were grown as previously described(Reference Giros, Grzybowski and Sohn19) and maintained at 37°C in a humidified atmosphere containing 5 % CO2. All cell lines were specifically bought for the present study from the American Type Culture Collection and none were used with more than fifty passes or 4 months. Cell cultures were tested monthly for Mycoplasma colonisation with LookOut™ Mycoplasma PCR Detection Kit (Sigma-Aldrich, St Louis, MO, USA).
Table 2 Several characteristics of five different colorectal cancer cell lines and status of genes of interest

MSI, microsatellite instability; MSS, microsatellite stability; APC, adenomatous polyposis coli; CTNNBI, cadherin-associated protein β1; KRAS, V-ki-ras2 kirsten rat sarcoma; MSH, Muts protein homolog; Cox-2, cyclo-oxygenase-2.
For experiments, cells were plated and allowed to adhere overnight. For preparation of whole cell lysates, 4 000 000 cells were used per 10 mm cell culture dish. In all other experiments, 200 000 cells were used per well of sixteen-well plate or dual chamber slide. Fermented samples were prepared for supplementation by centrifugation to separate solid material and filtration of the supernatant using a 0·22 μm filter. Cells were supplemented with faecal suspensions. These were set to contain a butyrate level of 4 mm for the PO treatment cells, which resulted in a level of 19 mm-propionate and 33 mm-acetate. In order to maintain the same concentration of the supernatant in the culture medium for the control cells, this resulted in SCFA levels of 0·8 mm-butyrate, 3·8 mm-propionate and 6·6 mm-acetate.
Experiments to assess apoptosis by 4′,6-diamidino-2-phenylindole (DAPI) or cytometry were performed in all cells 24 h post-supplementation. Detection of mitochondrial membrane depolarisation was done 18 h after treatment in all cell lines. Caspase activity was assessed in all cell lines 8 and 18 h post-supplementation. Gene expression experiments were performed after 2, 4, 8 and 12 h of PO treatment in all cell lines. Protein expression was detected by Western blotting in HCT116 and LoVo cells after 8 and 18 h treatment.
Apoptosis assessment by 4′,6-diamidino-2-phenylindole
Morphological assessment of apoptosis was performed 24 h post-supplementation with DAPI staining and mounted with Vectashield® mounting medium (Vector Laboratories, Inc., Burlingame, CA, USA) as previously described(Reference Llor, Pons and Roca20). Slides were visualised under a fluorescence microscope and apoptosis was assessed according to morphological criteria: cell shrinking, membrane blebbing, formation of apoptotic bodies and chromatin condensation.
Apoptosis quantification by flow cytometry
Cells were trypsinised 24 h post-supplementation, washed with 1 × PBS and stained with annexin-V/propidium iodine with Annexin-V-FLUOS (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer's instructions. Apoptosis was quantified with a BD FACSCalibur flow cytometer (San Jose, CA, USA). Apoptosis was also assessed after the addition of several caspase inhibitors: z-VAD-FMK (broad-spectrum caspase inhibitor); Ac-DEVD-CHO (caspase-3 specific inhibitor); Ac-IETD-CHO (caspase-8 specific inhibitor); and Ac-LEHD-CHO (caspase-9 specific inhibitor), all from BIOMOL International (Plymouth Meeting, PA, USA) in order to determine the implication of the different apoptosis pathways.
Preparation of whole cell protein lysates
Lysates were prepared as previously described(Reference Giros, Grzybowski and Sohn19). Briefly, 8 million cells were collected by scraping with 1 × PBS and lysed with 200 μl of lysis buffer. Incubation was carried out with agitation for 40 min at 4°C. Finally, cells were centrifuged at 16 000 g for 10 min at 4°C and the supernatant was collected. Protein concentration was measured by the Bradford assay (BioRad Laboratories, Hercules, CA, USA). Experiments were performed 8 and 18 h post-supplementation.
Caspase activity assay
Caspase activity was measured in whole cell lysates by fluorometric assay using DEVD-AMC, IETD-AMC and LEHD-AMC (Alexis Biochemicals, San Diego, CA, USA) as substrates for caspase-3, 8 and 9, respectively. A total of 75 μg of protein lysate were incubated with 10 μm of substrate in assay buffer (50 mm-HEPES pH 7·5, 20 % sucrose, 0·2 % 3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate, 10 mm-dithiothreitol) at 37°C. Caspase substrate cleavage releases 7-amino-4-methylcoumarin, which emits a fluorescent signal with 380 nm excitation and 460 nm emission. Fluorescence was registered from start of incubation for 3 h at 1 min intervals using a bonSAI fluorescence reader (Siemens, Deerfield, IL, USA). Fluorescence was calibrated using standard curves for 7-amino-4-methylcoumarin concentration. Experiments were performed 8 and 18 h post-supplementation.
Determination of mitochondrial membrane depolarisation
Cells were stained with the mitochondrial membrane potential marker JC1 (Sigma-Aldrich) for 1 h at 37°C and at a 4 μm concentration as previously described(Reference Giros, Grzybowski and Sohn19). Cells were then rinsed with medium and afterwards, fresh medium was added to the plates. Mitochondrial membrane potential was determined with fluorescence microscopy. Non-depolarised mitochondria display an orange colour and depolarised mitochondria display a green colour. Experiments were performed 18 h post-supplementation.
Determination of gene expression
Total RNA was extracted with RNAqueous®-4PCR (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions at 2, 4, 8 and 12 h post-supplementation. RNA (1 μg) was reverse-transcribed to complementary DNA using a High-Capacity complementary DNA Reverse Transcription kit (Applied Biosystems). TaqMan Low Density MicroFluidic cards (Applied Biosystems) were loaded with 50 ng of complementary DNA plus 50 μl of TaqMan gene expression master mix, per well. A total of four replicates were assayed for each gene: apoptosis-inducing factor (AIF), apoptotic protease activating factor 1 (APAF1), B-cell lymphoma protein 2 (Bcl2), Bcl-2-like protein 4 (Bax), B-cell lymphoma-extra large (Bcl-xl), Bcl-2 interacting domain (Bid), X-linked inhibitor of apoptosis (XIAP), Survivin FLICE-inhibitory protein (FLIP), second mitochondria-derived activator of caspases (Smac), Apo-1 (PAS), PAS-associated factor 1 (FAF1), Fas-associated death domain (FADD), PPARr (PPARG), cyclo-oxygenase-2, receptor interacting protein (RIP), TNF-α, death receptor (DR) 4, DR5, TNF receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL), TNF receptor type 1-associated DEATH domain protein (TRADD) and endogenous control (TFRC). Real-time PCR was performed with an ABI Prism 7900 (Applied Biosystems). Results were adjusted to endogenous control and normalised to experimental control. Relative expression (respiratory quotient, RQ) was calculated with the RQ manager program (Applied Biosystems). RQ values were calculated based on the comparative CT method(Reference Pfaffl21). Graphs reflect the plotted RQ and a relative expression of 1 means equal expression as control supplemented cells. ANOVA was used to assess statistically significant differences. P values of < 0·05 were considered statistically significant.
Protein expression analysis
Protein expression was assessed as previously described(Reference Giros, Grzybowski and Sohn19). Briefly, proteins from whole cell protein lysates were separated on a 10–12 % SDS-PAGE and transferred to Immobilon™ PVDF membranes (Millipore, Billerica, MA, USA). Membranes were blocked and then incubated for 1 h with the primary antibody against B-actin (Cell Signaling Technology, Danvers, MA, USA). The overnight incubation was carried out with the following primary antibodies: Bcl-xL, Bid, XIAP (Cell Signaling Technology), Bcl-2 homologous antagonist killer (Bak) and Bax (Sigma-Aldrich). Incubation with secondary antibodies was carried out for 1 h with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Cell Signaling Technology). Antibody binding was detected with SuperSignal West Pico (Pierce Biotechnology), and fluorescence was recorded on a blue sensitive X-ray film (Henry Schein, Melvile, NY, USA). Blot density was analysed with ImageJ 1.44 (NIH, http://rsbweb.nih.gov/ij/index.html) using 8-bit gel images with background subtracted under standard settings of 50·0 pixels rolling bar radius.
Statistical analysis
All experiments were repeated at least three times and each experiment included at least three replicates per condition. Statistical analysis was performed using the Kruskall–Wallis one-way ANOVA by rank and the Mann–Whitney U test to assess significant differences in apoptosis. Statistical significance was assumed with a P ≤ 0·05.
Results
Generation of SCFA after stool fermentation
The production of SCFA was approximately 1/5 in stool with no addition of PO as opposed to the stool where PO was added. Table 1 shows SCFA concentrations of acetic, propionic, isobutyric, butyric, isovaleric, valeric and caproic acids in stool with no added PO and 72 h after PO addition.
Apoptosis induction by Plantago ovata husk's fermentation products
Apoptosis induction was assessed by DAPI staining and quantified by flow cytometry. DAPI staining showed an important decrease in the number of cells treated with PO in relation to control cells. The remaining cells displayed clear apoptotic features, such as condensed chromatin, membrane blebbing and apoptotic bodies (Fig. 1(a)). Quantification by flow cytometry showed a significant increase in the percentage of apoptotic cells of between 1·8 × in Lovo cells and 4·7 × in SW480 cells (Fig. 1(b)). We then proceeded to examine different mechanisms leading to apoptosis in the CRC cells studied.

Fig. 1 (a) Plantago ovata husk (PO) fermentation products induced apoptosis. The panel shows a representative 4′,6-diamidino-2-phenylindole staining of LoVo and Caco-2 cells after 24 h treatment with PO. Orange arrows point to apoptotic bodies with condensed chromatin typical of cells undergoing apoptosis. (b) Flow cytometry quantification of apoptosis induction by PO fermentation products. The graph represents apoptosed cells as percentage of the total number of cells 24 h after supplementation of HCT116, LoVo, SW480, Caco-2 and HT-29 cells with Control (![]() ), PO fermentation products (
), PO fermentation products (![]() ) and PO+broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (zVAD) (
) and PO+broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (zVAD) (![]() ). All PO-treated cells exhibited significantly greater levels of apoptosis v. control treatment, while PO+zVAD treatment resulted in a much lower apoptosis induction in all cells though it didnot reach statistical significance in LoVo. Values are means with their standard errors represented by vertical bars. * Mean values were significantly different (P < 0·05). (c) Treatment with PO fermentation products induced caspase activity. Graphs represent specific caspase activity in pmol/min per μg of protein, 8 h (
). All PO-treated cells exhibited significantly greater levels of apoptosis v. control treatment, while PO+zVAD treatment resulted in a much lower apoptosis induction in all cells though it didnot reach statistical significance in LoVo. Values are means with their standard errors represented by vertical bars. * Mean values were significantly different (P < 0·05). (c) Treatment with PO fermentation products induced caspase activity. Graphs represent specific caspase activity in pmol/min per μg of protein, 8 h (![]() ) and 18 h (
) and 18 h (![]() ) after PO supplementation in HCT116, LoVo, SW480, Caco-2 and HT-29 cells. Results are corrected for the baseline caspase activity values present in control cells. Values are means with their standard errors represented by vertical bars. * Mean values were significantly different from those of control (P < 0·05). Caspase 3, 8 and 9 activity was significantly greater at 18 h compared to control in all cell lines.
) after PO supplementation in HCT116, LoVo, SW480, Caco-2 and HT-29 cells. Results are corrected for the baseline caspase activity values present in control cells. Values are means with their standard errors represented by vertical bars. * Mean values were significantly different from those of control (P < 0·05). Caspase 3, 8 and 9 activity was significantly greater at 18 h compared to control in all cell lines.
Plantago ovata-induced apoptosis is caspase-dependent. Implication of the intrinsic and extrinsic pathways
In order to establish the implication of the different caspase pathways, we quantified caspase activity using a fluorimetric assay. We assessed caspase-3, an executor caspase that is activated by both the intrinsic and the extrinsic pathways; caspase-8, the crucial initiator caspase of the extrinsic pathway, and caspase-9, essential in the activation of the intrinsic pathway. As shown in Fig. 1(c), the activity of all three caspases increased dramatically 18 h after supplementation with PO fermentation products in all the cells analysed, suggesting that both the intrinsic and extrinsic pathways are implicated in the pro-apoptotic effect of PO. The implication of caspases was further confirmed as the addition of a broad-spectrum caspase inhibitor z-VAD-FMK resulted in a significant decrease in the percentage of apoptotic cells in all but LoVo cells (Fig. 1(b)).
Implication of the B-cell lymphoma protein 2 family and activation of the mitochondrial pathway of apoptosis
The intrinsic pathway of apoptosis induction depends on a shift in the balance of pro- and anti-apoptotic members of the Bcl-2 family of proteins(Reference Schwarz, Andrade-Navarro and Gross22). Bax and 8 do have a pro-apoptotic effect and through their oligomerisation they permeabilise the mitochondrial outer membrane with the subsequent irreversible apoptotic cascade. On the other hand, anti-apoptotic elements such as Bcl-xL or Bcl-2 could prevent further oligomerisation and membrane permeabilisation functioning as defective constituents of Bax–Bak oligomers(Reference Leber, Lin and Andrews23). As shown in Fig. 2, control cells showed a decrease in Bak protein expression that was significant at 8 and 18 h in HCT116 cells and at 8 h in LoVo cells in comparison to PO-treated cells. On the other hand, PO-treated cells had significantly lower levels of anti-apoptotic Bcl-xL protein at 8 and 18 h in HCT116 and at 18 h in LoVo cells, indicating a clear shift towards a pro-apoptotic balance. A decrease in Bcl-xL gene expression was significant in all five cell lines except in Caco-2 cells (Table 3). No significant changes were seen in Bax protein expression in HCT116 cells while LoVo cells' expression was undetectable. No significant changes in protein expression were seen in Bid. Finally, Bcl-2 protein expression was not detected in any of the cell lines studied (data not shown).

Fig. 2 Plantago ovata husk (PO) treatment resulted in a shift towards a pro-apoptotic state of the B-cell lymphoma protein 2 (Bcl-2) family of proteins. The figure shows immunoblots of Bcl-2 homologous antagonist killer (Bak), Bcl-2-like protein 4 (Bax), Bcl-2 interacting domain (Bid) and Bcl-extra large (Bcl-xL) after treatment of HCT116 and LoVo cells with PO and control (C) at 8 and 18 h. Actin was used as housekeeping control protein. Control cells showed a decrease in Bak protein expression that was significant at 8 and 18 h in HCT116 cells and at 8 h in LoVo cells in comparison to PO-treated cells. The expression of pro-apoptotic Bak was increased at 18 h in HTC116, and at 8 h in LoVo cells. The expression of anti-apoptotic Bcl-xL was decreased compared to control at 8 and 18 h after PO treatment in HCT116 and at 18 h in LoVo cells. Bid and Bax did not show any expression changes, but the latter was undetectable in LoVo cells. Blot density was analysed with ImageJ 1.44 (NIH, http://rsbweb.nih.gov/ij/index.html) using 8-bit gel images with background subtracted under standard settings of 50·0 pixels rolling bar radius. Values are means with their standard errors represented by vertical bars. ![]() , PO 8h;
, PO 8h; ![]() , C 8h,
, C 8h, ![]() , PO 18h;
, PO 18h; ![]() , C 18h.
, C 18h.
Table 3 Gene expression changes of different elements involved in apoptosis regulation after treatment with Plantago ovata husk (PO) fermentation products in five colorectal cancer cell lines

AIF, apoptosis-inducing factor; APAF, apoptotic protease activating factor; Bax, B-cell lymphona-2-like protein 4; Bcl2, B-cell lymphoma protein 2; Bcl-xL, Bcl-extra large; BID, Bcl-2 interacting domain; XIAP, X-linked inhibitor of apoptosis; FLIP, FLICE-inhibitory protein; Smac, second mitochondria-derived activator of caspases; FADD, Fas-associated death domain; FAF1, FAS-associated factor 1; FAS, Apo-1; PPARG, PPARr; Cox-2, cyclo-oxygenase-2; RIP, receptor interacting protein; DR, death receptor; TNFR1, TNF receptor 1; TRAIL, TNF-related apoptosis-inducing ligand; TRADD, TNF-α receptor 1-associated death domain protein.
* Fold changes demonstrating a significant difference between PO-treated cells and control-treated cells.
† Low expression indicates mRNA targets which required greater than or equal to forty PCR cycles for detection.
The final result of a balance towards a pro-apoptotic environment results in mitochondrial membrane permeabilisation and subsequent release of key elements that trigger the apoptotic cascade. All PO-supplemented cells underwent mitochondrial membrane permeabilisation as assessed by JC1 stain, a mitochondrial potential marker. Control cells displayed mitochondria stained in red–orange, whereas PO-treated cells had a distinct green coloration clearly establishing induction of depolarisation by PO (Fig. 3(a)). Presumably as a consequence of the depolarisation, APAF1 expression was increased in all cells after PO treatment though it did not reach significance in Caco-2. The expression of AIF was increased in metastatic Lovo, HT-29 and Caco-2 cells though it was not significant in the latter. To the contrary, AIF was down-regulated in HCT116. Smac/Diablo was up-regulated in the same cells as AIF, reaching significance also in Caco-2 but not in SW480. On the other hand, the IAP Survivin was down-regulated in all cells after PO treatment, reaching significance in LoVo and HCT116. HT29 had undetectable levels. Finally, the inhibitor of apoptosis protein XIAP gene expression was down-regulated in HCT116, SW480 and HT-29, while there was a late up-regulation in LoVo cells that was not detected at the protein level (Fig. 3(b) and (c); and Table 3).

Fig. 3 (a) Mitochondrial membranes showed depolarisation 18 h after treatment with Plantago ovata husk (PO) fermentation products. Pictures show cells after JC-1 stain, a mitochondrial potential marker stain. Control cells show an orange colouration, indicating polarisation of the mitochondrial membrane, and PO-treated cells show a green colouration, as a result of mitochondrial membrane depolarisation. (b) Gene expression of intrinsic pathway regulators after treatment with PO fermentation products. Graphs represent variation of mRNA expression 2, 4, 8 and 12 h after PO supplementation, in respect to the control. Graphs reflect the plotted respiratory quotient (RQ) and a relative expression (RE) of 1 means equal expression as control supplemented cells. RQ value was calculated by the ΔΔCT method (endogenous control used was the TFRC or transferrin receptor gene). ANOVA was used to assess statistically significant differences. Values are means with their standard errors represented by vertical bars. Mean values were considered statistically significant different (P < 0·05). LoVo cells (![]() ) and HCT116 cells (
) and HCT116 cells (![]() ). Intrinsic pathway activators apoptosis-inducing factor (AIF), apoptotic protease activating factor 1 (APAF1) and second mitochondria-derived activator of caspases(Smac)/Diablo were up-regulated in PO-treated LoVo cells. Only APAF1 was significantly increased in HCT116 after PO. Anti-apoptotic Survivin was significantly down-regulated in both cell lines while X-linked inhibitor of apoptosis (XIAP) was down-regulated in HCT116 cells. (c) Regulation of anti-apoptotic XIAP protein by PO fermentation products. The figure shows immunoblots of XIAP 8 and 18 h after PO treatment and control (C). Actin was used as housekeeping protein. XIAP protein expression was down-regulated in HCT116 cells after PO treatment. LoVo cells had low XIAP protein expression.
). Intrinsic pathway activators apoptosis-inducing factor (AIF), apoptotic protease activating factor 1 (APAF1) and second mitochondria-derived activator of caspases(Smac)/Diablo were up-regulated in PO-treated LoVo cells. Only APAF1 was significantly increased in HCT116 after PO. Anti-apoptotic Survivin was significantly down-regulated in both cell lines while X-linked inhibitor of apoptosis (XIAP) was down-regulated in HCT116 cells. (c) Regulation of anti-apoptotic XIAP protein by PO fermentation products. The figure shows immunoblots of XIAP 8 and 18 h after PO treatment and control (C). Actin was used as housekeeping protein. XIAP protein expression was down-regulated in HCT116 cells after PO treatment. LoVo cells had low XIAP protein expression.
Implication of cellular death receptors
In order to determine the potential implication of cellular death receptors in the induction of apoptosis by PO, we studied the gene expression of FAS, DR5, DR4, TNF-R1, as well as TRADD, TNFα and RIP 2, 4, 8 and 12 h post-supplementation.
All cell lines showed rapid up-regulation of DR5 (2 h post PO supplementation) and a significant down-regulation of DR4, Fas and also TNFR1, although the latter was not significant in LoVo and Caco-2 cells (Fig. 4 and Table 3). On the other hand, while HCT 116 did not express TNF-α, only metastatic Lovo cells showed a significant increase of TNF-α expression, as early as 2 h post PO supplementation, accompanied by a significant up-regulation of two of the key elements of the death-inducing signalling complex (DISC): TRADD and RIP. Finally, FLIP was down-regulated after PO supplementation in all CRC cell lines, though it did not reach significance in metastatic Lovo cells (Fig. 4 and Table 3).

Fig. 4 Gene expression of extrinsic pathway regulators after treatment with Plantago ovata husk (PO) fermentation products. Graphs represent variation of mRNA expression 2, 4, 8 and 12 h after PO supplementation, respect to the control. Graphs reflect the plotted respiratory quotient (RQ) and a relative expression of 1 means equal expression as control supplemented cells. RQ value was calculated by the ΔΔCT method (endogenous control used was the TFRC or transferrin receptor gene). ANOVA was used to assess statistically significant differences. Values are means with their standard errors represented by vertical bars. Mean values were considered statistically significant different (P < 0·05) LoVo cells (![]() ) and HCT116 cells (
) and HCT116 cells (![]() ). Death receptor (DR) 5 was significantly up-regulated as early as 2 h after PO treatment, while other receptors DR4, Fas and TNF receptor 1 (TNFR1) had decreased expression. TNF-α (ligand) was dramatically up-regulated in metastatic LoVo cells, but no significant expression was detected in HCT116 cells. Death-inducing signalling complex (DISC) members receptor interacting protein (RIP) and TNF receptor type 1-assocaited DEATH domain protein (TRADD) were up-regulated only in LoVo cells. Finally, the key extrinsic pathway inhibitor FLICE-inhibitory protein (FLIP) had decreased expression in HCT116 only.
). Death receptor (DR) 5 was significantly up-regulated as early as 2 h after PO treatment, while other receptors DR4, Fas and TNF receptor 1 (TNFR1) had decreased expression. TNF-α (ligand) was dramatically up-regulated in metastatic LoVo cells, but no significant expression was detected in HCT116 cells. Death-inducing signalling complex (DISC) members receptor interacting protein (RIP) and TNF receptor type 1-assocaited DEATH domain protein (TRADD) were up-regulated only in LoVo cells. Finally, the key extrinsic pathway inhibitor FLICE-inhibitory protein (FLIP) had decreased expression in HCT116 only.
Discussion
The main objectives of the present study were: to assess if PO fibre supplementation can induce CRC cell apoptosis, and to investigate the mechanisms implicated in this anti-neoplastic effect. Apoptosis is one of the crucial anti-carcinogenic mechanisms by which damaged or mutated cells are eliminated. For this purpose, we used Plantaben®, which contains PO, a mucilaginous fibre that had been associated with a reduction in CRC risk(Reference Juarranz, Calle-Puron and Gonzalez-Navarro14) and mortality(Reference Lopez, Villanueva and Martinez-Hernandez15). Fermentation was carried out in anaerobic conditions using human faecal microbiota to mimic in vivo conditions and all products generated by this process were used to supplement cells and assess apoptosis induction. Therefore, cell supplements contained high concentrations of propionic and acetic, besides butyric acid. While butyric acid is well known to induce colonocyte apoptosis, to our knowledge only another recent study by Borowicki et al. (Reference Borowicki, Stein and Scharlau24) has addressed this effect using all SCFA generated through anaerobic fibre fermentation.
Our present study results have shown that there is a significant induction of apoptosis in all CRC cells after supplementation with the fermentation products of PO. Apoptosis induction is independent of p53, APC, cadherin-associated protein β1 (β-catenin), mismatch repair or cyclooxygenase-2 expression status, as the studied cells have different levels of alteration in regards to these genes (Table 2), which suggests that this beneficial effect of PO is not limited to specific colorectal carcinogenic pathways and it could be potentially useful as an anti-carcinogenic agent in a wide group of CRC. PO-induced apoptosis is mediated by caspases. Caspase 3, as well as caspase 8 and 9, activities are increased, demonstrating involvement by both the extrinsic and intrinsic pathways.
The involvement of the intrinsic pathway was further confirmed by the shift in the balance towards an increase in pro-apoptotic Bak expression and a decrease in anti-apoptotic Bcl-xL in both HCT116 and LoVo cells, in line with results previously reported in butyrate-treated Caco-2 cells(Reference Roy, Dionne and Marx25, Reference Ruemmele, Schwartz and Seidman26). The final result of a balance towards a pro-apoptotic environment results in mitochondrial membrane permeabilisation(Reference Rasola and Bernardi27). Permeabilisation allows inter-membrane space proteins, including caspase activators (cytochrome-C and Smac/Diablo) as well as caspase-independent death effectors (AIF), to be released into the cytosol. Cytochrome-C binds to APAF1 and mediates its conformational change facilitating the activation of the initiator caspase-9 in a direct fashion via the assembly of the apoptosome causing the activation of effector caspases. Smac/Diablo favours the caspase cascade indirectly by antagonising the activity of endogenous inhibitors of caspases, such as IAP(Reference Galluzzi, Zamzami and de La Motte Rouge28). In all PO-treated cell lines, we observed a depolarisation of the mitochondrial membrane along with a subsequent increase of caspase activator Smac/Diablo and death effector AIF expression in most cell lines, except in HCT116, presumably due to their release after mitochondrial depolarisation. We also observed a significant increase in the apoptosome member APAF1 expression and a decrease in the inhibitors of apoptosis Survivin and XIAP expression, except in LoVo cells where there was a late increase in XIAP expression. Derangements of these intrinsic pathway components have well documented important clinical consequences. For example, decreased expression of APAF1 in CRC correlates with tumour progression and aggressive phenotype(Reference Paik, Jang and Song29, Reference Zlobec, Minoo and Baker30). Survivin over-expression in CRC has been associated with worse prognosis(Reference Xiaoyuan, Longbang and Jinghua31), higher risk of relapse(Reference Yie, Lou and Ye32) and local recurrence(Reference Rodel, Hoffmann and Distel33), while Survivin down-regulation is key in the apoptosis induction of some chemotherapeutic agents(Reference Liu, Hu and Chao34, Reference Kaneko, Tsuji and Asanuma35). XIAP expression has been correlated to poor clinical outcome and therapy resistance(Reference Danson, Dean and Dive36), but XIAP inhibition can increase sensitivity to some chemotherapeutic agents(Reference Shrikhande, Kleeff and Kayed37, Reference Lacasse, Kandimalla and Winocour38) and was found to be useful in the treatment of chemo-refractory cancers(Reference Cillessen, Hess and Hooijberg39). As down-regulation of these important apoptosis inhibitors had not been seen in a previous study on butyrate-treated cells(Reference Ruemmele, Schwartz and Seidman26), we cannot rule out that this could be related to the presence of the other fatty acids or even totally distinct products generated through PO fermentation. All together, PO's regulation of crucial aspects of the intrinsic pathway of apoptosis suggests important anticancer effects.
Several members of the TNF receptor family and their ligands are key elements in the induction of apoptosis via death receptors. Some of these receptors, such as TrailR2 (or DR5), TrailR1 (or DR4), Fas or TNFR1, contain death domains in their cytosolic tails. The activation of these receptors results in receptor clustering and recruitment of intracellular proteins, such as TRADD and RIP, to the cytosolic domains forming the DISC that triggers caspase activation and leads to apoptosis(Reference Wallach, Varfolomeev and Malinin40). On the other hand, there is evidence that some of these receptors, namely DR4 and TNFR1, engage in parallel pathways and besides apoptosis induction they can also send an anti-apoptotic signal through the activation of the NF-κB pathway(Reference Chen, Fu and Raja41). FLIP inhibits caspase-8 activation, blocking the apoptotic signal transduction through the extrinsic pathway. In the present study, the extrinsic pathway of apoptosis was also induced in PO-treated cells. That is, most cells showed up-regulation of death receptor DR5, while Fas and DR4 showed significant down-regulation. TNFR1 was also significantly down-regulated in HCT116, HT-29 and SW480. As DR4 and TNFR1 may alternatively activate the pro-survival NFkB pathway(Reference Chen, Fu and Raja41), down-regulation by PO could further promote cell death by suppressing survival signals. Finally, DR4 over-expression has been associated with worse disease-free and overall survival in some CRC patients(Reference van Geelen, Westra and de Vries42).
While death receptors DR4, DR5 and Fas were similarly regulated by PO in all cell lines, other important extrinsic pathway elements were differentially regulated in metastatic v. non-metastatic cells. As such, the remarkable up-regulation of TNF-α ligand along with DISC components RIP and TRADD in metastatic LoVo cells suggests a distinctive mechanism of apoptosis activation. This could be potentially important, as a higher endogenous expression of TNF-α has been associated with decreased metastatic potential and better prognosis in CRC(Reference Fukushima, Tanizaki and Takagi43–Reference Takagi, Tomita and Fukushima45).
In primary tumour but not metastatic CRC cell lines, the extrinsic pathway modulator FLIP was significantly down-regulated after PO treatment (Table 3). FLIP may inhibit both spontaneous death ligand-independent and death receptor-mediated apoptosis in CRC cells(Reference Wilson, McLaughlin and McEwan46). FLIP competes with pro-Caspase-8 and -10 for binding to FADD, thus squelching death receptor signalling. Several tumours, including CRC, contain inappropriately elevated levels of FLIP, rendering them resistant to apoptosis induction by death domain signalling. Interestingly, several important chemotherapeutic agents have been shown to induce apoptosis through down-regulation of FLIP expression(Reference Roue, Perez-Galan and Lopez-Guerra47–Reference Day, Najafi and Wu49), which suggests an important anti-carcinogenic mechanism of PO in CRC.
In summary, using a setting that mimics physiological conditions in the gut, we have shown that supplementation with PO induces apoptosis in different types of CRC cells. This induction is mediated by both the intrinsic and extrinsic apoptosis pathways and is the result of modulating important regulators of these pathways. This regulation involves some distinct elements in primary tumour v. metastatic CRC cells. The presented results suggest that PO husk could potentially be useful as an adjuvant element in chemotherapy treatments.
Acknowledgements
The present research was supported by grant no. 2007-01934 from Rottapharm S. L. and Departmental grants from the University of Illinois at Chicago Department of Medicine and Cancer Center. All authors contributed to the preparation of the paper and agreed with the content of the submitted manuscript. V. R. S., A. G., R. M. X., L. F., M. G. and R. M. X. performed the research. V. R. S., A. A. and X. L. analysed the data. X. L. wrote the paper. All authors declare that there are no conflicts of interest.







