


Epidemiological studies examining the relationship between folate and colon cancer indicate that diminished folate status, as measured by dietary folate intake or by blood folate levels, is associated with an increased risk of colon cancer(Reference Choi and Mason1). Low folate status is also associated with the risk of hepatocellular carcinogenesis in humans(Reference Kuo, Lin and Wu2, Reference Welzel, Katki and Sakoda3). However, a temporal increase in colon cancer rate observed after nationwide folic acid fortification programmes suggests that a high folate intake might increase the risk of colon cancer(Reference Hirsch, Sanchez and Albala4–Reference Sauer, Mason and Choi6), especially among the elderly who may already have a precursor of cancer or cancer-prone conditions. A recent double-blind, placebo-controlled, randomised clinical trial with older subjects has demonstrated that a high-dose folic acid supplementation was associated with a higher risk of having three or more colonic adenomas and prostate cancer(Reference Cole, Baron and Sandler7). The biological mechanism underlying the effect of high-dose folate supplementation on carcinogenesis has not yet been elucidated.
Since folate is a critical co-enzyme for de novo nucleotide synthesis(Reference Choi and Mason1, Reference Jang, Mason and Choi8), folate depletion reduces thymidylate levels in the deoxyribonucleotide pool, increasing uracil misincorporation into DNA(Reference Blount, Mack and Wehr9). Uracil misincorporation is sensitive to changes in folate status and is regarded as a biomarker of folate status(Reference Basten, Duthie and Pirie10). Recent observations in animal studies have demonstrated that folate depletion results in an excess of uracil misincorporation into DNA(Reference Choi, Friso and Dolnikowski11, Reference Linhart, Troen and Bell12). Many studies also suggest that excess uracil in DNA causes abnormalities in the DNA and chromosomal structure. DNA repair mechanisms can excise uracil residues, but the repair process temporarily produces an abasic site and a break in the DNA strand. Double-strand breaks result when transient strand breaks are closely spaced on the opposite strands of DNA, and this can increase the chances of translocations, rearrangements, deletions and duplications that can activate proto-oncogenes or inactivate tumour suppressor genes(Reference Choi and Mason1, Reference Blount and Ames13, Reference Fenech, Aitken and Rinaldi14). Uracil misincorporation is thus thought to be the biological mechanism explaining how folate affects carcinogenesis(Reference Choi and Mason1).
An alternative pathway for thymidylate synthesis does not rely on folate. Thymidylate can be produced from thymidine using the enzyme thymidine kinase. In cases of decreased de novo synthesis due to folate depletion or anti-folate treatment, the cell uses this salvage pathway to produce thymidylate(Reference Choi, Shane and Selhub15). Our previous animal study found that supplementation of thymidine partially restores the DNA repair process that was impaired due to folate depletion in colonic cells(Reference Choi, Kim and Weitzel16). However, no study to date has demonstrated whether dietary supplementation of thymidine reduces uracil misincorporation under conditions of reduced de novo thymidylate synthesis. Under normal conditions, 2–5 % of dietary thymidine is incorporated into nucleic acids via the salvage pathways, confirming the bioavailability of oral thymidine. Metabolic stressors including trauma, rapid growth and limited food supply increase the conversion of dietary nucleotides into nucleic acids(Reference Adjei, Yamauchi and Chan17).
The purposes of the present study are to determine whether uracil misincorporation into DNA is associated with ageing and dietary folate levels in the colon and liver, and to determine whether thymidine supplementation can reverse the increased uracil misincorporation that is induced by low dietary folate.
Materials and methods
Animals and diets
The present study on folate and ageing was reviewed and approved by the Institutional Animal Care and Use Committee of the USDA Human Nutrition Research Center on Aging at Tufts University. Forty-two weanlings and forty-two 18-month-old male C57BL/6 mice were used. Old mice were 22·5 months old at the conclusion of the study, an age that is considered to be true murine senescence. The diets are amino acid-defined diets described by Walzem & Clifford(Reference Walzem and Clifford18). For 20 weeks, the mice received four different diets with three different concentrations of dietary folate: folate deplete (0 mg/kg diet); folate replete (2 mg/kg diet); folate supplemented (8 mg/kg diet); folate deplete (0 mg/kg diet) with thymidine supplementation (1·8 g/kg diet). The basal requirement of folate for mice is 2 mg/kg, and 1·8 g/kg of thymidine have been used to evaluate the preventive effect of nucleoside–nucleotide supplementation on colonic mucosal damage in mice and rats(Reference Adjei, Yamauchi and Chan17). Individually housed mice were group pair-fed to decrease the variability in food and nutrient intakes within and between dietary groups. All mice in each age group were given a measured portion of food daily. The amount of food supplied to each dietary group was matched to the mean daily food consumption of the group within that age category with the least food consumption. The mice were killed at 20 weeks, and liver and colon samples were harvested as described previously(Reference Choi, Friso and Dolnikowski11).
Liver folate concentration
The concentrations of folate in the diet and liver tissue were determined by a microtitre plate assay using the Lactobacillus casei assay(Reference Tamura, Picciano, Stokstad and Gregory19).
Uracil misincorporation into DNA
Uracil content was measured in both colonic and liver DNA. Uracil in DNA was measured by a previously described GC/MS method(Reference Choi, Friso and Dolnikowski11). Uracil was removed from 15 μg DNA by incubating with 4 U uracil DNA glycosylase for 2 h at 37°C. After the glycosylase digestion, 100 pg of labelled uracil (13C4H4O215N; Cambridge Isotope Laboratories, Cambridge, MA, USA) were added as an internal standard, and the samples were dried in a speed vacuum concentrator. The residue was resuspended in a mixture of 50 μl acetonitrile, 10 μl triethylamine and 1 μl 3,5-bis-(trifluoromethyl)-benzyl bromide. This mixture was shaken to derivatise for 25 min at 30°C, and 50 μl water (HPLC grade) were added to improve the extractability. N 1, N 3-(3,5-bis-[trifluoromethyl]-benzyl)uracil, a derivatised form, was extracted into 100 μl iso-octane. Then, 5 μl of the sample were injected into a Series 6890 gas chromatograph equipped with a 7683 automated liquid sampler (Agilent, Palo Alto, CA, USA). The splitless mode was utilised with a purge activation time of 1 min, and a split vent was achieved on an Ultra 2 capillary column (Hewlett Packard). A 5973 network mass selective detector (Agilent) using negative chemical ionisation MS was used for analysis: selected ion monitoring at m/z 337 and 343. The amount of uracil in DNA was presented as pg uracil/μg DNA.
Statistics
The primary analytical method employed in the present study is a two-way ANOVA to examine the effect of ageing as well as of diets. Post hoc analyses with the least significant difference test were conducted to reduce the incidence of type I error and to determine the locations of differences after ANOVA. A P value of < 0·05 is the criterion for significance in all instances. As uracil data (pg/μg) were skewed, log transformation was performed to achieve normality in order to meet the assumptions of parametric statistical tests. Data are presented as the means and standard deviations.
Results
Animals
During the 20-week feeding period, six old mice died: one from the folate-deplete group; one from the folate-deplete, thymidine-supplemented group; four from the folate-replete group. Deaths were attributed to liver cancer or unknown causes. None of the young mice died. Overall, weight changes were not different between each dietary group within the same age group (data not shown).
Liver folate concentration
Mean liver folate concentrations (μg/g tissue) increased with increased dietary folate in both young and old mice (Table 1). The thymidine supplementation (1·8 g/kg diet) provided to the folate-deplete group did not affect liver folate concentrations in either the young or old mice group (data not shown).
Table 1 Liver folate concentrations in old (18 months) and young (4 months) male C57BL/6 mice fed the folate-deplete (FD) diet (0 mg/kg), folate-replete (FR) diet (2 mg/kg) and folate-supplemented (FS) diet (8 mg/kg)
(Mean values and standard deviations)

a,b,c Mean values within a row with unlike superscript letters were significantly different (comparison by the least significant difference test, P < 0·05).
Uracil misincorporation into colon and liver DNA according to differences in folate status
Skewed data were log-transformed for analysis, but data shown in each table are non-transformed values. In the old mice, uracil content in colonic DNA was not different among the different dietary folate groups (Table 2). At 20 weeks, of the eight mice in the young folate-supplemented group, three showed very high uracil levels in colonic DNA, resulting in a wide variance in values. Nevertheless, uracil content in the colonic DNA was significantly higher in the folate-supplemented group than in the folate-deplete and -replete groups (P < 0·05) (Table 2).
Table 2 Uracil in the colonic DNA of old and young male C57BL/6 mice fed the folate-deplete (FD) diet (0 mg/kg), folate-replete (FR) diet (2 mg/kg) and folate-supplemented (FS) diet (8 mg/kg)*
(Mean values and standard deviations)

a,b Mean values within a row with unlike superscript letters were significantly different (comparison by the least significant difference test, P < 0·05).
* All data were log-transformed for comparison.
In the liver, the mean value of uracil misincorporation into DNA was higher in the folate-deplete group than in the folate-replete and folate-supplemented groups among the old mice, but the difference did not reach a statistically significant level (P < 0·1; Table 3). In young mice, uracil misincorporation into liver DNA was not different among the different folate groups. Within the same dietary folate group, there was no difference in uracil misincorporation into DNA between the young and old mice as well as between the liver and the colon.
Table 3 Uracil in the liver DNA of old and young male C57BL/6 mice fed the folate-deplete (FD) diet (0 mg/kg), folate-replete (FR) diet (2 mg/kg) and folate-supplemented (FS) diet (8 mg/kg)*
(Mean values and standard deviations)

a,b Mean values within a row with unlike superscript letters were significantly different, comparison by the least significant difference test (different tendency P < 0·1).
* All the data were log-transformed for comparison.
Uracil misincorporation into colon and liver DNA in the thymidine-supplemented group
In old mice, there was less uracil misincorporation into liver DNA in the thymidine-supplemented group than in the folate-deplete group (P = 0·027, comparison by the least significant difference test) (Fig. 1). However, in both old and young mice, uracil misincorporation into colonic DNA was not different between the folate-deplete group and the thymidine-supplemented, folate-deplete group.
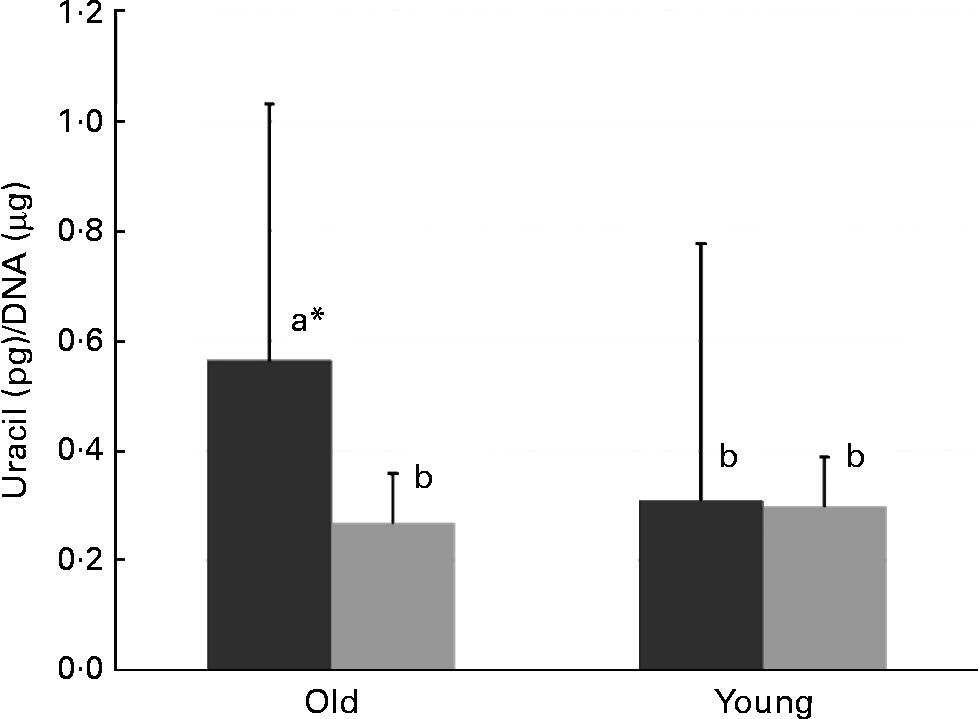
Fig. 1 Uracil misincorporation into the liver DNA of the folate-deplete (FD, ■) and FD, thymidine-supplemented (![]() ) groups. * Values in old mice (P = 0·027). a,b Mean values with unlike letters were significantly different (P < 0·05; comparison by the least significant difference test).
) groups. * Values in old mice (P = 0·027). a,b Mean values with unlike letters were significantly different (P < 0·05; comparison by the least significant difference test).
Discussion
In the present study, we demonstrated different levels of uracil misincorporation into DNA according to tissue type, age and dietary folate levels. Our observations provide an important mechanistic consideration for the possibility that mandatory folate fortification and voluntary folate supplementation may increase the risk of colon cancer. Interestingly, folate supplementation (8 mg/kg, four times higher than the basal requirement) increases uracil misincorporation into colonic DNA compared with the folate-replete and -deplete groups in young mice, while there was no difference among the different dietary folate groups in the colon of aged mice (22 months old). This result is not consistent with previous studies, which have demonstrated that a folate-deficient diet increased uracil misincorporation into colonic DNA in older Sprague–Dawley rats (16 months old)(Reference Choi, Friso and Dolnikowski11) and uracil DNA glycosylase-deficient (Ung − / − ) young mice(Reference Linhart, Troen and Bell12). However, the present results are consistent with a folate intervention trial with 5 mg folic acid and 1·25 mg vitamin B12. van den Donk et al. (Reference van den Donk, Pellis and Crott20) demonstrated that high-dose folate and vitamin B12 supplementation increases uracil misincorporation into DNA of rectal biopsy samples from subjects (aged 18–80 years) who had colorectal adenomas. The present study is also consistent with the most recent folate supplementation trial by Hazra et al. (Reference Hazra, Selhub and Chao21), demonstrating a lack of changes in DNA-uracil in rectal biopsy samples from older adults who were supplemented with 1 mg folate/d for 3 years.
We previously proposed that a high folate status by folic acid supplementation may inhibit one-carbon metabolism and thereby increase colonic carcinogenesis(Reference Sauer, Mason and Choi6). Increased genomic uracil in the colon of young, folate-supplemented mice observed in the present study supports the hypothesis that folic acid supplementation may inhibit the thymidylate synthesis pathway in the colon, and that the young mice might be more sensitive to this type of inhibition than the aged mice. Thus, we can speculate that increased uracil misincorporation into colonic DNA through a high intake of folic acid in young individuals might be able to initiate colonic carcinogenesis through its mutagenic effect from a young age.
In contrast to colonic DNA, even though it did not reach a statistically significant level, liver DNA demonstrated a stepwise decrease in mean values of DNA-uracil in parallel with the incremental increase in dietary folate from folate-deplete to folate-supplemented in aged mice (P < 0·1), but not in young mice. This result is opposite to that of the colon. However, similar observations are found in other randomised controlled trials using blood DNA. Folic acid supplements decreased uracil misincorporation into DNA using the RDA level, 400 μg(Reference Kapiszewska, Kalemba and Wojciech22), and a level three times higher than RDA, 1·2 mg(Reference Basten, Duthie and Pirie10). Also, the tissue specificity of one-carbon metabolism is well known. One-carbon metabolism in the liver is especially different from other organs that have different enzyme activities. For example, homocysteine in the liver is metabolised by three distinct pathways: folate-dependent remethylation using methionine synthase; folate-independent remethylation using betaine:homocysteine methyltransferase; or trans-sulfuration using cystathionine-β-synthase. In contrast, in the colon, enzymes involved in the trans-sulfuration and folate-independent remethylation pathways have lower activities compared with liver(Reference Bauchart-Thevret, Stoll and Burrin23). Furthermore, liver and colon tissues have different isoenzymes of methionine adenosyltransferases(Reference Chen, Xia and Lin24). Thus, the different responses of one-carbon metabolism to folate supplementation between the liver and the colon are not surprising. It appears that folate supplementation may inhibit one-carbon metabolism in the colon but not in the liver where folate can be stored.
Folate depletion seems to increase uracil in the liver DNA of the aged mice. In a rodent model, methyl-deficient diet including low folate increased the development of liver tumours. Epigenetic mechanisms, including DNA methylation, are suggested due to the decreased availability of the methyl donor(Reference Mikol, Hoover and Creasia25, Reference Zapisek, Cronin and Lyn-Cook26). In this regard, increased uracil misincorporation into liver DNA could also be a link between folate deficiency and hepatocellular carcinogenesis(Reference Yu, Yuan and Lu27). Thus, reduced thymidylate synthesis due to folate depletion could be one responsible mechanism for liver tumour development in the methyl-deficient diet rodent model of hepatocellular carcinoma.
In the present study, thymidine supplementation reduced uracil misincorporation into liver DNA. Thymidylate (deoxythymidine 5′-monophosphate, 5-methyluracil-2′-deoxyriboside-5′-phosphate) is an essential pyrimidine for DNA synthesis that is produced through two pathways: a folate-dependent de novo pathway and a folate-independent salvage pathway(Reference Kisliuk28). Thymidylate is produced de novo from deoxyuridine-5′-monophosphate (deoxyuridine) by the action of the folate-dependent enzyme thymidylate synthase. Additionally, thymidylate can be salvaged from thymine deoxyriboside (thymidine) by the action of thymidine kinase. Thymidylate synthesis using the salvage pathway is increased with folate depletion or in stress conditions that require significantly more DNA synthesis.
There was no difference in uracil misincorporation into liver DNA between the young and old mice in the same dietary folate groups. Even though liver folate concentrations are similar between the young and old mice, it appears that uracil misincorporation is correlated with dietary folate in aged but not in young mice. Furthermore, thymidine supplementation reduces uracil misincorporation only in the aged mice. These observations suggest that uracil content in liver DNA in old mice is correlated with an inadequate synthesis of thymidylate due to low dietary folate, while the young mice might adequately compensate for the increment of uracil by folate depletion. We can explain this discrepancy using two different mechanisms. First, ageing alters the distribution of folate forms and may reduce the amount of folate used for thymidylate synthesis. We previously reported the effect of ageing on folate distribution in one-carbon metabolism(Reference Choi, Friso and Dolnikowski11). Second, ageing can reduce the removal and repair of uracil misincorporation into DNA(Reference Xu, Herzig and Rotrekl29). Dietary folate can affect uracil status not only through thymidylate synthesis but also through the process of uracil removal and repair. A most recent rat study demonstrated that folate deficiency alters O6-methylguanine-DNA methyltransferase and 8-oxoguanine-DNA glycosylase DNA repair protein expression in the liver and colon, indicating that folate status can significantly modulate DNA repair(Reference Duthie, Grant and Pirie30).
In conclusion, the effects of folate and thymidine supplementation on uracil misincorporation into DNA are tissue and age specific. Since increased uracil misincorporation into DNA may affect carcinogenesis, further studies are needed to clarify the significance of the increase in uracil misincorporation into colonic DNA of folate-supplemented young mice.
Acknowledgements
The present study was supported by the US Department of Agriculture, under agreement no. 581950-9-001. Any opinions, findings, conclusion or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the US Department of Agriculture. The authors have no conflicts of interest. This project has been supported in part by the National Institute of Health Grants R21 AA016681 and R01 AG025834 (S.-W. C.). All authors participated in the conception, design and conduction of the study as well as in the interpretation of the data and drafting the manuscript. K.-C. K., H. J. and S.-W. C. also performed the generation, collection and analysis of the data.




