


As early as the 1960s, the relevance of the monocyte–lymphocyte ratio (MLR) for disease prediction was pointed out. The first studies focused on infectious diseases, suggesting MLR reflects the balance between effector and host (Hanifin & Cline, Reference Hanifin and Cline1970), and found MLR to predict the development and progress of tuberculosis (Iqbal et al., Reference Iqbal, Umbreen and Zaidi2014). In later studies, the association between MLR and other diseases was studied and the MLR emerged as a predictor for cancer (Nishijima et al., Reference Nishijima, Muss, Shachar, Tamura and Takamatsu2015). While MLR is examined in relation to disease, studies examining the causes of variance in MLR in the general population are lacking. To fully understand the role of MLR in disease, it is, however, necessary to understand the factors underlying variation in MLR in the general non-patient population.
We recently showed that heritability plays a role in individual differences in two other lymphoid ratios, the neutrophil–lymphocyte ratio (NLR) and the platelet–lymphocyte ratio (PLR). For NLR, the heritability was moderate (35%), but for PLR, heritability was high (64%), with evidence of the presence of non-additive genetic effects (Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016). The first genome-wide association study (GWAS) for these two ratios identified a genome-wide locus on the HBS1L-MYB intergenic region associated with PLR (Lin et al., Reference Lin, Montoro, Bell, Boomsma, de Geus and Jansen2016), which has been associated with multiple blood parameters, including platelet count (Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016).
Although the heritability of MLR has not been studied, genetic studies have been conducted for its subcomponents, the monocyte and lymphocyte levels, showing heritability estimates of 56–73% for monocyte levels and 35–66% for lymphocyte levels (Evans et al., Reference Evans, Frazer and Martin1999; Hall et al., Reference Hall, Ahmadi, Norman, Snieder, MacGregor and Vaughan2000). In addition, linkage and GWAS have pointed to the genetic variants partly responsible for the individual variation in monocyte and lymphocyte levels. GWASs have identified eight loci associated with monocyte levels: ITGA4 at 2q31.3, HLA-DRB1 at 6p21.32, CCBP2 at 3p22.1, RPN1 at 3q21.3, LPAR1 at 9q31.3, intergenic regions at 8q24 and 3q21, and IRF8 at 16q24.1 (Crosslin et al., Reference Crosslin, McDavid, Weston, Zheng, Hart and Andrade2013; Keller et al., Reference Keller, Reiner, Okada, van Rooij, Johnson and Chen2014; Nalls et al., Reference Nalls, Couper, Tanaka, van Rooij, Chen and Smith2011; Reiner et al., Reference Reiner, Lettre, Nalls, Ganesh, Mathias and Austin2011). GWAS also identified two loci for lymphocyte levels: 6p21 (EPS15L1 gene) and 19p13 (Crosslin et al., Reference Crosslin, McDavid, Weston, Zheng, Hart and Andrade2013; Nalls et al., Reference Nalls, Couper, Tanaka, van Rooij, Chen and Smith2011). Taken together, genetic factors are likely to play a role in normal variation in MLR, but the nature of the involvement remains to be determined.
Environmental and lifestyle factors may also influence MLR levels. Here, too, studies on MLR itself are lacking, but our own study on PLR and NLR (Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016) showed that lifestyles influence PLR and NLR levels, and agreed with other studies (Azab et al., Reference Azab, Camacho-Rivera and Taioli2014; Li et al., Reference Li, Chen, Luo, Hong, Pan and Lin2015) that smoking and BMI may also affect these parameters. In addition, its subcomponents, monocyte and lymphocyte levels, have been found to be influenced by BMI (Tenorio et al., Reference Tenorio, Farah, Ritti-Dias, Botero, Brito and Moura2014; Zaldivar et al., Reference Zaldivar, McMurray, Nemet, Galassetti, Mills and Cooper2006) and smoking (Schwartz & Weiss, Reference Schwartz and Weiss1994), although these effects are not found in all studies (Al-Sufyani & Mahassni, Reference Al-Sufyani and Mahassni2011; Perez-de-Heredia et al., Reference Perez-de-Heredia, Gomez-Martinez, Diaz, Veses, Nova and Warnberg2015).
In this article, we examined several potential causes of variance in MLR in the general population. First, age, sex, smoking, and BMI effects on MLR were investigated. Then, we studied genetic influences on MLR to provide more insight into its genetic architecture in a healthy population. We started by estimating the heritability of MLR and it subcomponents by extended twin family modeling. Next, we used GWAS to identify genetic variants associated with MLR variation and genome-wide complex trait analysis (GCTA) to determine the percentage of variance of MLR that is explained by significant versus all measured genetic variants (single nucleotide polymorphisms; SNPs). Subsequently, we performed an expression quantitative trait loci (eQTL) analysis of all the top SNPs, which were significantly associated with MLR. We repeated the GWAS, GCTA, and eQTL analyses for monocyte levels, while referring to lymphocyte levels in the results presented previously (Lin et al., Reference Lin, Montoro, Bell, Boomsma, de Geus and Jansen2016). Finally, linkage disequilibrium (LD) score regression was performed using the summary statistics of the GWAS results to determine the polygenetic effects and genetic correlations between MLR and subcomponents.
Methods
Participants and Phenotypes
All participants were adults registered with the Netherlands Twin Register (NTR), who took part in a longitudinal study on health and lifestyle in twins and their family members (Willemsen et al., Reference Willemsen, Vink, Abdellaoui, den Braber, van Beek and Draisma2013). Data were obtained as part of NTR biobanking projects conducted in 2004–2008 (Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks and Estourgie-van Burk2010, Reference Willemsen, Vink, Abdellaoui, den Braber, van Beek and Draisma2013; Sirota et al., Reference Sirota, Willemsen, Sundar, Pitts, Potluri and Prifti2015). After removing outliers (i.e., absolute values exceeding mean ±5 SD), data on monocyte and lymphocyte count were available for 9,501 participants clustered in 3,412 families. During the interview conducted at the time of the home visit, height and weight were obtained. BMI was calculated as weight (kg) divided by height squared (m2). Participants reported whether they currently smoked or had smoked. If so, they were asked for the number of cigarettes smoked per day and how long they (had) smoked. Participants were divided into five categories: non-smoker, ex-smoker, light smoker (currently smoking less than 10 cigarettes a day), average smoker (currently smoking 10–19 cigarettes a day), and heavy smoker (currently smoking 20 or more cigarettes a day). Participants were asked to indicate when they were last ill and the nature of the illness. In the case of medication use, the dosage, brand, and name were recorded. In addition, participants indicated on the presence and nature of any chronic disease. The following exclusion criteria were used to identify individuals who may have had a compromised immune system at the time of blood sampling: (1) illness reported in the week prior to sampling (N = 552); (2) C-reactive protein (CRP) ≥ 15 (N = 307); (3) basophile count > 0.02 × 109/L (N = 154); (4) report of blood-related disease or cancer (N = 84); and (5) use of anti-inflammatory medication (N = 423), glucocorticoids (N = 143), or iron supplements (N = 29). Participants meeting one or more of these criteria were labeled as unhealthy (N = 1,362), leaving 8,139 individuals from 3,280 families as the population that we will here refer to as the healthy population. Genetic twin-family modeling was conducted using data from twin families limited to at most one twin pair per family and at most two brothers and two sisters and father and mother. This resulted in a sample of 7,513 participants from 3,252 families, including 240 monozygotic male (MZM), 98 dizygotic same-sex male (DZM), 536 monozygotic female (MZF), 219 dizygotic same-sex female (DZF), and 222 dizygotic opposite-sex (DOS) twin pairs. The study protocol was approved by the Medical Ethics Committee of the VU University Medical Center Amsterdam and all participants provided informed consent.
Procedure
Participants were visited at home, or when preferred, at work, to obtain blood samples and conduct a brief health-related interview. Visits took place in the morning between 7 am and 10 am and women were seen, when possible, between the 2nd and the 4th day of the menstrual cycle or, if on hormonal birth control, were visited in their pill-free week. Participants were asked to fast from the evening before and to refrain from smoking or physical exercise 1 hour before blood sampling (for more details see Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks and Estourgie-van Burk2010). Peripheral blood was collected in anticoagulant vacuum tubes, which were inverted 8–10 times immediately after the blood draw. All samples were transported to the laboratory facility in Leiden, the Netherlands, within 3–6 hours after blood sampling. The blood samples were then directly used or stored to measure parameters of interest or extract DNA or RNA at a later moment (see Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016; Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks and Estourgie-van Burk2010). The hematological profile was obtained from EDTA blood samples with a Coulter system (Coulter Corporation Miami, USA). This profile consisted of total white blood cell count, percentages and numbers of neutrophils, lymphocytes, monocytes, eosinophils, and basophils, and indicators of red blood cell types and platelets. We calculated MLR as the absolute monocyte count (109/L) divided by the absolute lymphocyte count (109/L). CRP was determined from a heparin plasma sample using the 1,000 CRP assay (Diagnostic Product Corporation).
Genotype Data
For DNA isolation, we used the GENTRA Puregene DNA isolation kit on frozen whole blood samples, which were collected in EDTA tubes. All procedures were performed according to the manufacturer's protocols (Boomsma et al., Reference Boomsma, Willemsen, Sullivan, Heutink, Meijer and Sondervan2008). Genotyping was done on multiple platforms, including a number of partly overlapping subsets of participants. The following platforms were used: Affymetrix Perlegen 5.0, Illumina 370, Illumina 660, Illumina Omni Express 1 M, and Affymetrix 6.0 (for details see (Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016). The individual SNP markers were lifted over to Build 37 (HG19) of the Human Reference Genome using the LiftOver tool (http://genome.sph.umich.edu/wiki/LiftOver). Genotype calls were made with platform specific software (BIRDSUITE APT-Genotyper Beadstudio) for each specific array. Phasing of all samples and imputing cross-missing platform SNPs was done with MACH 1 (Li et al., Reference Li, Willer, Ding, Scheet and Abecasis2010). The phased data were then imputed with MINIMAC (Howie et al., Reference Howie, Fuchsberger, Stephens, Marchini and Abecasis2012) in batches of around 500 individuals for the autosomal genome using the above 1000G Phase I integrated reference panel for 561 chromosome chunks obtained by the CHUNKCHROMOSOME program (Liu et al., Reference Liu, Li, Wang and Li2013). SNPs were removed if the Mendelian error rate was > 0.02, if the imputed allele frequency differed more than 0.15 from the 1000G reference allele frequency, and if MAF < 0.01 and if R2 < 0.80. Hardy–Weinberg Equilibrium was calculated on the genotype probability counts for the full sample and SNPs were removed, if the p value <.00001. After imputation, MZ twins were reduplicated back into the data. This left 6,010,458 SNPs in the GWAS analyses.
As several different platforms were used, additional SNP quality control (QC) included an evaluation of the SNP platform effects, and SNPs showing platform effects were removed. This was done by defining individuals on a specific platform as cases and the remaining individuals as controls. Allelic association was then calculated and SNPs were removed if the specific platform allele frequencies were significantly different from the remaining platforms with p value <.00001. In total, 5,987,253 SNPs survived this QC and these SNPs were then used to build the genetic relationship matrix (GRM) for all individuals. The selected SNPs were transformed to best guess Plink binary format and subsets were made for each of the 22 chromosomes. The GRMS for all NTR samples were then calculated using GCTA (Yang et al., Reference Yang, Lee, Goddard and Visscher2011). We generated 24 GRMs in total. A first autosomal GRM reflects an identity-by-state (IBS) matrix for all individuals. This GRM is determined from all autosomal SNPs and is used to estimate the SNP heritability (h2 g). A second autosomal GRM represents closely related individuals (identity-by-state [IBS] > 0.05), so any remaining pairwise relationship estimates smaller than 0.05 were set to zero in this matrix. This matrix is used as second covariate matrix in the GWAS and GCTA studies to account for the family structure of individuals and to estimate the narrow-sense heritability (h2) of applying an additive model. Finally, 22 GRMs were created that included all autosomal SNPs except for those on the one chromosome that is tested in the GWAS (the leave one chromosome out or LOCO strategy). These matrices were used in the GWASs as a covariate matrix to remove artificial inflation due to all kinds of subsample stratification.
Statistical Analyses
First, using age- and sex-corrected values, we tested for differences in MRL, monocyte, and lymphocyte levels between the healthy and unhealthy population using a t-test. Next, within the healthy population, we explored the age and sex effects by linear regression. To detect the influence of lifestyle on variation in the MLR level, we included BMI and smoking behavior in a regression analysis conducted separately by sex, taking age into account. Analyses were conducted in STATA (Stata Corp., 2013) using the cluster option to correct for the family structure within the data. Using genetic structural equation modeling in OpenMx (Boker et al., Reference Boker, Neale, Maes, Wilde, Spiegel and Brick2011), the heritability of MLR, monocyte count, and lymphocyte count was estimated in the healthy population. MLR, monocyte count, lymphocyte count, and age were standardized using z scores. Parameters were estimated by maximum likelihood. We summarized the family resemblance with respect to MLR by means of correlations corrected for age, sex, and age × sex effects. Then, we fitted a series of genetic models. The total phenotypic variance was decomposed into four sources of variation: additive genetic (A), non-additive genetic (D), common environmental (C), and unique environmental (E) variation. The common environmental variance reflects the variance shared between siblings and twins (VC). The resemblance among family members was modeled as a function of A, D, and C. We allowed for a correlation in phenotype between spouses (µ). In fitting the genetic models, we included as covariates age, sex, sex × age. We fitted the full model as described and tested the presence of assortative mating (i.e., the correlation between phenotypes of spouses) and the presence of shared environment and non-additive influences. The nested submodels were compared to the full model by log likelihood ratio test (-2LL) using a significance level of 0.05.
GWAS
We performed two GWASs: one for MLR level and one for monocyte count, using the quality controlled imputed SNPs, including age, sex, three Dutch principal components (PCs) generated with the Eigensoft software and genotype platform as covariates (N = 5,892; see Methods and Abdellaoui et al., Reference Abdellaoui, Hottenga, de Knijff, Nivard, Xiao and Scheet2013). As we had already conducted a GWAS for lymphocyte count, using a largely overlapping sample (N = 5, 901, overlap of 5,890 individuals) we did not rerun this analysis but instead refer here to the results published in Lin et al. (Reference Lin, Montoro, Bell, Boomsma, de Geus and Jansen2016). Analyses were performed with the GCTA software running a mixed linear model association model to account for relatedness (Tucker et al., Reference Tucker, Loh, MacLeod, Hayes, Goddard and Berger2015). To avoid inflated test statistics in datasets with related individuals and other remaining cryptic stratification, we used two covariate GRM matrices: the matrix for all individuals, excluding the chromosome under analysis (LOCO analysis) and the matrix only focusing related individuals with IBS > 0.05 (Tucker et al., Reference Tucker, Loh, MacLeod, Hayes, Goddard and Berger2015). For the GWAS, we assumed the statistically significant threshold as a p value less than 5 × 10−8 (Pe'er et al., Reference Pe'er, Yelensky, Altshuler and Daly2008), and we refer to as marginally significant when p values exceed this threshold but remain below 10−4.
eQTL Analysis
To detect possible causal effects for significant genetic variants, we conducted eQTL analyses, in which eQTL effects were detected with a linear model approach using MatrixeQTL (Shabalin, Reference Shabalin2012). The analysis specified expression level as the dependent variable and SNP genotype values as the independent variable. The eQTL dataset is described in detail elsewhere (Jansen et al., in press; Wright et al., Reference Wright, Sullivan, Brooks, Zou, Sun and Xia2014). eQTL effects were defined as cis when the distance between probe set–SNP pairs was smaller than 1M base pairs (Mb), and as trans when the SNP and the probe set were separated by more than 1 Mb on the genome according to hg19.
GCTA and Linkage Disequilibrium Score Regression
We performed GCTA analyses to estimate narrow-sense heritability, the fraction of genetic variance explained by the significant SNPs detected in the GWAS and the fraction of genetic variance explained by the known significant SNPs from the published literature. These analyses were done for MLR level, monocyte count, and lymphocyte count. A restricted maximum likelihood analysis procedure was used under a linear design (Yang et al., Reference Yang, Lee, Goddard and Visscher2011). Sex, age, genotype platform, and three Dutch PCs were included as covariates. We used two covariance matrixes to estimate narrow sense heritability (h2), and GWAS and known loci heritability (Zaitlen et al., Reference Zaitlen, Kraft, Patterson, Pasaniuc, Bhatia, Pollack and Price2013). The first GRM is the full autosomal GRM as described previously. The second GRM is the closely related (IBS > 0.05) matrix. Pearson correlations between the phenotypes of interest were calculated in R (R Core Team., 2014). Whether polygenetic effects (Bulik-Sullivan et al., Reference Bulik-Sullivan, Loh, Finucane, Ripke, Yang and Patterson2015) influenced MLR and its compositions was explored by LD score regression. The SNP heritability (Finucane et al., Reference Finucane, Bulik-Sullivan, Gusev, Trynka, Reshef and Loh2015) of MLR, monocyte count, lymphocyte count, and genetic correlations (Bulik-Sullivan et al., Reference Bulik-Sullivan, Loh, Finucane, Ripke, Yang and Patterson2015) among the phenotypes were determined by LD score regression on our computed GWAS summary statistics. The genetic correlation of two traits can be calculated by the slope from the LD regression on the product of effect sizes (z score) for two phenotypes of interest. In order to do this, we used the HapMap3 LD scores (N SNPs = 1,293,150) computed for each SNP based on the LD observed in European ancestry individuals from the 1,000 Genomes project (accessible online at http://github.com/bulik/ldsc). QC for genetic data is the default setting in the program.
Results
Health Status, Sex, Age, and Lifestyle
Table 1 gives the descriptive statistics for MLR and its subcomponents, the monocyte and lymphocyte count, for the healthy and unhealthy parts of the population. The comparison of the healthy and unhealthy population (see Methods for definition), taking sex and age into account as well as family structure, showed, as expected, that individuals in the unhealthy population had on average a higher MLR ratio, t(9,499) = −7.95, p < .001, and monocyte count, t(9,499) = −5.06, p < .001, and a lower lymphocyte count, t(9,499) = −2.57, p = .01). We continued our investigation in the healthy population, examining the influence of age and sex. Men had higher MLR levels than women, b = −0.0176, p < .01, and MLR increased with age in both men and women, b = 0.0013, p < .001. There was also evidence for an age × sex interaction: the age effects were alleviated in the women. With respect to the subcomponents, monocyte, and lymphocyte levels were higher in men than in women and increased with age. These age effects were similar in men and women. To test the effects of BMI and smoking, we included this variable in a regression analysis conducted separately by sex and taking age into account. The results, shown in Table 2 (model 1), indicate that smoking is related to a decrease in MLR level in both men and women. BMI was not associated with MLR in either sex. However, an age × BMI interaction was seen for MLR in women (model 2): the age effects were alleviated by an increased BMI level. The BMI and smoking effects were also examined in the MLR subcomponents: Higher BMI and being a smoker were related to higher monocyte and lymphocyte levels. For lymphocyte count in women, there was evidence for an age × BMI interaction, again indicating a reduction of the BMI effect at an older age.
TABLE 1 Average Levels (SD) for MLR and Its Subcomponents in the Healthy and Unhealthy Population for Men and Women
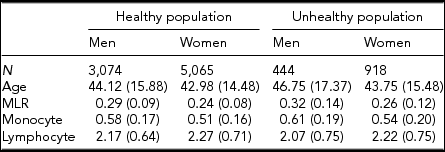
TABLE 2 Results of the Linear Regression Analyses (Regression Coefficients) for MLR and Its Subcomponents for Men and Women

Estimates in bold are significant (*p < .05, **p < 0.01, ***p < .001). MLR = monocyte count ratio; BMI = body mass index; Age × BMI = interaction of age and BMI; Age × smoking = interaction of age and smoking; Model 1: blood variable = b0 + b1×age + b2×BMI + b3×smoking; Model 2: blood variable = b0 + b1×age + b2×BMI + b3×smoking + b4×age×BMI + b5×age×smoking.
Heritability
The known genetic relations among mono- and di-zygotic twins and their family members were used to model familial resemblance in MLR, and monocyte and lymphocyte count as a function of genetic and environmental parameters. These models included sex, age, and sex × age effects as fixed effects. Table 3 contains the familial correlations as obtained for MLR, monocyte, and lymphocyte count. For MLR, twin pair correlations did not depend on sex, and the correlations did not differ across DZ twin and sibling relations. The correlations in MZ males and MZ female twin pairs were equal as were the other male and female first-degree relative correlations. The resulting MZ correlation was 0.43 (CI is 0.33–0.46) and the DZ correlation was 0.22 (0.14–0.24), with spousal correlations significant at 0.104 (0.002–0.135). The pattern of twin correlations showed no evidence for non-additive or common environmental effects. This was confirmed by model fitting in which the heritability of MLR was estimated at 40% (0.34–0.43).
TABLE 3 Familial Correlations (Confidence Interval) for MLR Monocyte and Lymphocyte Count Within the Healthy Population

Correlations in bold type were obtained from submodels in which all matching correlations of the tested subgroup of family relations were constrained to be equal.
We also conducted these series of genetic modeling analyses for monocyte and lymphocyte count. For monocyte count, there were no significant spousal correlations and the MZ correlation was 0.58 (0.54–0.62) while the DZ correlation was 0.27 (0.21–0.31). In line with the pattern of the correlations, genetic modeling estimated the broad sense heritability at 58%, with non-additive effects accounting for 12% and no evidence for the influence of common environmental factors. For lymphocyte count, we estimated the heritability in the current set (N = 5,892, with >99% overlap with the set described in Lin et al., Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016) and, as to be expected, results were similar to those published in Lin et al. (Reference Lin, Hottenga, Abdellaoui, Dolan, de Geus and Kluft2016) with a broad sense heritability at 58% and non-additive effects accounting for 22%.
GWAS
Figures 1, 2 and 3 show the QQ and Manhattan plots from the GWAS for MLR, monocytes and lymphocytes. After adjusting for age, sex, genotype platform, and PCs, and using the LOCO and family-based GRM correction, the GWAS λs were 0.9965 for MLR and 1.0166 for monocyte count.

FIGURE 1 Manhattan and QQ plot for MLR level with SNPs having a minor allele frequency above 0.01 (λ = 0.996503).

FIGURE 2 Manhattan and QQ plot for monocyte count with SNPs having a minor allele frequency above 0.01 (λ = 1.0166495).

FIGURE 3 Manhattan and QQ plot for lymphocyte count with SNPs having a minor allele frequency above 0.01 (λ = 1.022341).
For MLR, associations were found with 11 SNPs situated on the ITGA4 (VLA-4 subunit alpha) gene on chromosome 2q31 (Figure 1 and Table 4). The top SNP rs3755021 T allele was linked to a decrease in MLR level (β = −0.012, p = 2.21E-8). This SNP was not associated with lymphocyte count, but in our study was marginally significantly associated with monocyte count (β = −0.018, p = 6.34E-6), and has also been associated with monocyte count in a linkage study (Maugeri et al., Reference Maugeri, Powell, Hoen, de Geus, Willemsen and Kattenberg2011) and two previous GWA studies (Crosslin et al., Reference Crosslin, McDavid, Weston, Zheng, Hart and Andrade2013; Nalls et al., Reference Nalls, Couper, Tanaka, van Rooij, Chen and Smith2011). The G allele of rs6740847 in this region has been linked to decreased ITGA4 expression levels in blood, which increases the number of circulating monocytes and may indicate this is a causal gene (Maugeri et al., Reference Maugeri, Powell, Hoen, de Geus, Willemsen and Kattenberg2011).
TABLE 4 Significant SNP Associations for MLR

For monocyte count, the four top hits were rs13029501 at ITGA4, rs55929401 located at a region nearby LPAR1 at 9q31.3, rs391855 at IRF8, and rs9469532 at 6p21. The most significant locus rs13029501 at 9q31 has been previously associated with monocyte count in European and Japanese populations (Ferreira et al., Reference Ferreira, Hottenga, Warrington, Medland, Willemsen and Lawrence2009; Kamatani et al., Reference Kamatani, Matsuda, Okada, Kubo, Hosono and Daigo2010; Maugeri et al., Reference Maugeri, Powell, Hoen, de Geus, Willemsen and Kattenberg2011; Nalls et al., Reference Nalls, Couper, Tanaka, van Rooij, Chen and Smith2011). This SNP is 14.6kb upstream of the SNP most significant associated MLR level (rs3755021, the pair wise LD is 0.22). It is located in a region 163kb downstream of lysophosphatidic acid receptor 1 gene (LPAR1, also known as EDG2) and increases LPAR1 expression, which is linked to an increased number of monocytes (Maugeri et al., Reference Maugeri, Powell, Hoen, de Geus, Willemsen and Kattenberg2011). As indicated previously, genetic variants nearby the ITGA4 region are involved in the down regulation of ITGA4 expression, which increases the number of monocytes circulating in peripheral blood. The IRF8 gene has also been associated before with monocyte count and has been identified as multiple sclerosis susceptibility loci (De Jager et al., Reference De Jager, Jia, Wang, de Bakker, Ottoboni and Aggarwal2009). Animal model studies showed that IRF8 as a transcription factor plays an essential role in the regulation of lineage commitment during monocyte differentiation (Kurotaki et al., Reference Kurotaki, Osato, Nishiyama, Yamamoto, Ban and Sato2013; Terry et al., Reference Terry, Deffrasnes, Getts, Minten, van Vreden and Ashhurst2015; Yanez et al., Reference Yanez, Ng, Hassanzadeh-Kiabi and Goodridge2015). The top SNP at 6p21 rs9469532 is an intergenic genetic variant nearby ITPR3, LOC101929188, and LOC105375023. The HLA-DRB1 region 1,043kb upstream of this SNP has previously been associated with monocyte count (Okada et al., Reference Okada, Hirota, Kamatani, Takahashi, Ohmiya and Kumasaka2011).
As published in Lin et al. (Reference Lin, Montoro, Bell, Boomsma, de Geus and Jansen2016), there were no significant hits when conducting the GWAS for lymphocyte count. However, it is of interest to note that the locus on chromosome 6p21, which was associated with the monocyte level, was also marginally associated with MLR (for rs9469532, β = −0.069, p = 7.69E-5) and lymphocyte count (for rs114641912, β = −0.059, p = 6.19E-6). This region harbors candidate genes like ITPR3 (Oishi et al., Reference Oishi, Iida, Otsubo, Kamatani, Usami and Takei2008) and HLA-DRB1 (Farragher et al., Reference Farragher, Goodson, Naseem, Silman, Thomson and Symmons2008), which have been previously implicated in immunological diseases. In addition, other loci with ‘potential association peaks’, meaning p values are low but do not reach the required significance level, have been found to be associated with immune disease such as ERAP1 at 5q15 (Alvarez-Navarro et al., Reference Alvarez-Navarro, Martin-Esteban, Barnea, Admon and de Castro2015) and CNTN5 at 11q22 (Thomas et al., Reference Thomas, Gazouli, Karantanos, Rigoglou, Karamanolis and Bramis2014).
Table 5 shows the loci for monocyte and lymphocyte count found in previous studies and their significance levels for MLR and its subcomponents in the current study. For some loci, p values were low, indicating a ‘potential’ for association, even though they did not reach the required significance level. For example, rs9880192, located in the intergenic region between c3orf27 and rs1991866, an intergenic variant at 8q24.21, shows p values <10−6 for monocyte level and <10−3 for MLR.
TABLE 5 Known Blood Cell Count Loci and their Significance in Our GWAS

Chr = chromosome; p = p values from previous studies; p (mlr) = p values for MLR in current study; p (mono) = p values for monocyte count in current study; p (lymp) = p values for lymphocyte count in current study.
eQTL Results
Among the significant GWA loci for MLR and blood cell counts, there were a number of associations between the SNPs of interest and nearby gene expression (Table 6). However, the SNPs identified in our GWAS have low LD (r 2 < 0.8) with the top SNPs associated with gene expression, which suggest the GWAS SNPs are not part of the functional eQTL locus. Furthermore, no eQTLs with trans-effects were identified. In conclusion, we did not detect any cis- or trans-effects for the SNPs of interest.
TABLE 6 Overview of eQTL Results: The Association Between Genetic Variants of Interest (β) with Gene Expression Level, Uncorrected for Blood Composition

LD r 2: LD between GWAS SNP and top SNP in eQTL analysis. β = eQTL β of GWA SNP, FDR = eQTL FDR GWA SNP.
GCTA and LD Score Regression
The results of the GCTA are shown in Table 7. From the GCTA we found a narrow-sense heritability of 43.3% for MLR, 54.1% for monocyte count, and 51.7% for lymphocyte count. The significant SNPs obtained in the GWAS for MLR explained 0.6% of the variance in MLR, and the significant SNPS obtained in the GWAS for monocyte count explained 4.4% of the variance in monocyte count. All known loci from published literature together explained 1.3% of MLR variance, 2.4 % of monocyte count variance and 0.3% of lymphocyte count variance. In LD score regression, all λ values were larger than the LD score regression intercept, and intercepts were close to 1, indicating that the inflation of the p value distribution from the GWAS results is caused by polygenetic effects, rather than population stratification. The SNP heritability of MLR, monocyte count, and lymphocyte count when applying LD score regression was 13%, 17%, and 19%, respectively (see Table 8).
TABLE 7 Narrow Sense Heritability (Standard Error) and the Proportion of Genetic Variance Explained by Known Significant SNPs (Standard Error) According to GCTA Analyses for Monocyte Count Ratio and its Subcomponents

p values in bold type indicate significant estimates.
TABLE 8 LD Score Regression Results for MLR, Monocyte Count and Lymphocyte Count

Estimates for heritability and intercept with SE in brackets. All estimates are significant (p > 0.05).
In addition, we observed positive phenotypic correlations between MLR and monocyte count (r = 0.550, p < .0001) and between monocyte count and lymphocyte count (r = 0.386, p < .0001), and a negative phenotypic correlation between MLR and lymphocyte count (r = −0.494, p < .0001). However, despite the presence of phenotypic associations, no significant genetic correlations were detected between any pair of variables.
Discussion
We presented a detailed examination of the causes of variance in MLR in the general population. Health status was, as expected, an important determinant of MLR level: individuals with a compromised immune system, our so-called unhealthy group, had on average a higher MLR than the healthy participants. In the healthy population, smoking, sex, and age and their interactions were important determinants of variation in MLR and its subcomponents. Smoking was associated with a higher MLR and this is in line with the higher MLR in the individuals with a compromised immune system in our study and the higher MLR seen in cancer patients. Genetic factors also were shown to play a role in MLR variation in the general population. Heritability for MLR was estimated at 40% and MLR level was associated with a locus near ITGA4. Earlier studies have shown this locus to be associated with monocyte levels. Heritability estimates were higher for MLR subcomponents (58% for both lymphocyte and monocyte count) and, in contrast to MLR, there was evidence for non-additive effects. Monocyte level was also associated with ITGA4, and four more genes were related to monocyte level in our analyses, replicating findings in previous GWA studies. For lymphocyte level, no significant genetic variants emerged.
From these results, it is clear that the genetic variants associated with blood cell counts may also influence their balance, as reflected in their ratios. In addition to the genetic variants in ITGA4 genes, which were significantly associated with both MLR and monocyte count, there were a number of loci that were significantly associated with monocyte count that may also affected MLR level: the loci near LPAR1, IRF8, and ITPR3 were marginally significant associated with MLR level. Also, a locus near c3orf27 was marginally significantly associated with both MLR and monocyte count. We did not see evidence for pleiotropic genetic variants associated with MLR and lymphocyte count. To understand the role of the genetic variants in MLR variation, we investigated what is known about the role the identified genetic variants play in regulating gene expression. However, we did not find any evidence for cis-effects or trans-effects of these genetic variants.
Among three phenotypes, the narrow-sense heritability h2 of lymphocyte (35.3% in the healthy population) was the lowest, but its SNP heritability was the highest (19.12% from the LD score regression). These results suggest more common autosomal SNPs may be associated with lymphocyte count. The LD score regression results showed that polygenetic effects, rather than confounding factors, explain both ratio and count variance. Although there are significant overall correlations and an overlap in associated genetic variants has been detected between MLR and monocyte count, no significant genetic correlations were detected among variables, suggesting that the polygenetic effects are too small to be detected with the current sample size.
Overall, this series of studies provided more insight into the causes of variation in MLR within the general population. While the genetic pathways as well as non-genetic causes of variance still need more clarification, it is clear that these factors need to be taken into consideration when studying the relationship between MLR and disease development.
Acknowledgments
BD Lin is supported by a PhD grant (201206180099) from the China Scholarship Council. This study was supported by multiple grants from the Netherlands Organization for Scientific Research (NWO: 016-115-035, 463-06-001, and 451- 04–034), ZonMW (31160008 and 911-09-032); Institute for Health and Care Research (EMGO+) and Neuroscience Campus Amsterdam (NCA); Biomolecular Resources Research Infrastructure (BBMRI–NL, 184.021.007), European Research Council (ERC-230374); Biobanking and Biomolecular Resources Research Infrastructure (BBMRINL: 184.021.007). Genotyping was made possible by grants from NWO/SPI 56-464-14192, Genetic Association Information Network (GAIN) of the Foundation for the National Institutes of Health, Rutgers University Cell and DNA Repository (NIMH U24 MH068457-06), the Avera Institute, Sioux Falls (USA) and the National Institutes of Health (NIH R01 HD042157-01A1, MH081802, Grand Opportunity grants 1RC2 MH089951 and 1RC2 MH089995).











