



Introduction
Cryopreservation of human semen is used as a method for preserving fertility in various cases of male factor infertility 1-2 (Cavalla et al., Reference Cavalla, Rovei, Masera, Vercellino, Massobrio, Mutani and Revelli2006; Anderson, Reference Anderson2008). Negative effects of cryopreservation on fertilization capacity, motility and viability parameters of spermatozoa have been well documented in the literature (Yoshida et al., Reference Yoshida, Hoshiai, Fukaya, Ohi, Tozawa and Mandai1990; Nallella et al., Reference Nallella, Sharma, Allamaneni, Aziz and Agarwal2004; Ozkavukcu et al., Reference Ozkavukcu, Erdemli, Isik, Oztuna and Karahuseyinoglu2008). As a result, the use of frozen sperm cells in assisted reproductive techniques (ART) including intrauterine insemination (IUI), in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI), has been reported to reduce the outcomes of these treatments, therefore resulting in reduced fertilization rates, impaired developmental and implantation potential of embryos, and to an increase in abortion rates (Yildiz et al. Reference Yildiz, Ottaviani, Law, Ayearst, Liu and McKerlie2007). There is limited knowledge of the molecular basis of the cryopreservation process and sperm damage. The supposed mechanisms underlying the negative effects of freezing include membrane damage and alterations in both membrane permeability, structure of cellular skeleton and mitochondrial damage, plus DNA fragmentation, oxidative stress, capacitation and increase in sperm morphology (Royere et al., Reference Royere, Hamamah, Nicolle and Lansac1991; Parks & Graham, Reference Parks and Graham1992; Cormier et al., Reference Cormier, Sirard and Bailey1997; Watson, Reference Watson2000; Donnelly et al., Reference Donnelly, McClure and Lewis2001; Linfor & Mayers, Reference Linfor and Mayers2002; Peña et al., Reference Peña, Johannisson, Wallgren and Rodríguez Martınez2003; Chohan et al., Reference Chohan, Griffin and Carrell2004; Ortega-Ferrusola et al., Reference Ortega-Ferrusola, Sotillo-Galán, Varela-Fernández, Gallardo-Bolaños, Muriel, González-Fernández, Tapia and Peña2008; Ozkavukcu et al., Reference Ozkavukcu, Erdemli, Isik, Oztuna and Karahuseyinoglu2008). Damage that arises as a result of the freezing procedure is called as ‘cryodamage’ and may be due to intracellular and extracellular ice formation and osmotic stress during freezing and thawing processes (Mazur, Reference Mazur1984; Royere et al., Reference Royere, Hamamah, Nicolle and Lansac1991; Oehninger et al., Reference Oehninger, Duru, Srisombut and Morshedi2000; Stanic et al., Reference Stanic, Tandara, Sonicki, Simunic, Radakovic and Suchanek2000; Schoolcraft et al., Reference Schoolcraft, Moffatt, Sakkas, Seli and Gardner2004; Yildiz et al., Reference Yildiz, Ottaviani, Law, Ayearst, Liu and McKerlie2007; Said et al., Reference Said, Gaglani and Agarwal2010) which may cause apoptosis, a genetic process activated for self-suicide of the cell.
Apoptosis is defined as ‘programmed cell death’ coded by intrinsic factors. It is activated to eliminate old and damaged cells in an organism. It is not possible to distinguish an apoptotic sperm cell during routine microinjection procedures. Therefore, it is impossible to eliminate the damaged sperm cell after cryopreservation and this may cause impaired fertilization and embryo development, and reductions in embryo quality, pregnancy rates, implantation rates and also an increase in abortion rates.
Apoptosis is suggested to play an important role in the cryo-injury of sperm DNA, as the process of cryopreservation has been shown to increase the activation of specific aspartic acid-directed cysteine proteases (caspases) in both human (Paasch et al., 2004a, Wündrich et al., Reference Wündrich, Paasch, Leicht and Glander2006; Karabulut et al., 2014) and bull spermatozoa (Martin et al., 2007). The exact apoptotic mechanisms involved in the aetiology of sperm cryopreservation, including those leading to cryo-injury of sperm DNA, have not been fully understood.
Signs of apoptosis include phosphatidylserine degradation products, changes in mitochondrial membrane potential, and particularly in DNA fragmentation and the caspase cascade as a result of caspase activation. Caspases, are known to play key roles in the cellular apoptotic cascade and eventual cell death (Thornberry & Lazebnik, Reference Thornberry and Lazebnik1998). Apoptosis markers such as DNA fragmentation and caspase activations are observed at the end of the apoptotic process and this change is irreversible after those signs are detected. Activated caspases attack target molecules such as nucleic acids, proteins and lipids that lead to different and diverse outcomes in the cells such as DNA fragmentation and lipid peroxidation (Hetts, Reference Hetts1998).
Caspase-3 is an executioner caspase and is activated after cleaving by the initiator caspases. The ICAD–CAD (Inhibitors of Caspase-Activated DNase–Caspase-Activated DNase) complex is one of the substrates of caspase-3, and is excised by activated caspase-3. CAD is an endonuclease and it is set released and activated, and degrades chromosomal DNA leading to DNA fragmentation (Karp, Reference Karp2007).
The aim of the present study was to investigate the possible effects of sperm cryopreservation on main apoptotic signs including DNA fragmentation and caspase-3 activation, and to determine if these effects varied according to semen quality.
Methods
Study design
Cryopreserved sperm samples of 72 patients who attended the Florence Nightingale Hospital, IVF Center between January 2016 and June 2016 because of different indications (medical treatments, n = 8, surgical treatments, n = 32 chemotherapy or radiotherapy, n = 16 cryoptozoospermia, n = 16) were included in the study. Samples from smokers, patients with a history of varicocele, and patients with genetic abnormalities were not included in the study. Semen analysis was performed according to World Health Organization (1999) criteria. Briefly, semen concentration (million/ml), and total and progressive motile sperm rates (%) were assessed before and after cryopreservation. Normal morphology rate (%) were assessed according to Kruger’s strict criteria (Kruger et al., Reference Kruger, Acosta, Simmons, Swanson, Matta, Veeck, Morshedi and Brugo1987). Semen analyses were performed by the same technician. DNA fragmentation rates and caspase-3 activation levels were analyzed before and after the cryopreservation procedure.
Among the 72 participants, 39 were normozoospermic, and three had at least one abnormal sperm parameter including concentration, motility and morphology (<20 million/ml sperm concentration, <50% total motility) according to WHO-1999 criteria and 14% normal morphology according to Kruger’s strict criteria (Kruger et al., Reference Kruger, Acosta, Simmons, Swanson, Matta, Veeck, Morshedi and Brugo1987): <70% normal morphology) (oligoasthenoteratozoospermic: 28%; e: 3%; asthenoteratozoospermic: 2%).
Sperm cryopreservation and thawing
The liquid nitrogen vapour procedure was used to cryopreserve the samples. Sperm samples were collected in a sterile container following masturbation. Liquefaction semen analysis was performed according to WHO-1999 criteria. The cryoprotectant solution (TYB; Irvine Scientific, Santa Ana, CA, USA) was added (1:1 sample/cryoprotectant ratio) on the semen sample very slowly, and the samples were transferred cryotubes. The cryotubes were then placed 10–15 cm above the liquid nitrogen level and were exposed to liquid nitrogen vapour for 10 min until freezing, and then put directly into liquid nitrogen.
Frozen semen samples were thawed 7 days later by placing the cryotubes in a water bath at 30°C for 10 min until the solution melted completely. The samples were then left at room temperature for 20 min and washed with G-IVF (VitroLife, Sweden) medium. For this purpose, 1 ml of G-IVF is added to the solution which was then centrifuged at 1300 rpm for 10 min; the supernatant was discarded to get remove the cryoprotectant. This step was repeated two times and the pellet was used for examination.
Sperm DNA fragmentation assessment
The terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labelling (TUNEL) assay was used to analyse DNA fragmentation in sperm cells. DNA fragmentation was analysed both before and after the cryopreservation procedure. In Situ Cell Death Detection Kit, Fluorescein, Roche, was used for TUNEL procedure, which was performed as recommended in the datasheet.
The samples were centrifuged at 2000 rpm and were separated from the seminal plasma. The pellets were suspended three times in 1 ml of distilled water, homogenized and centrifuged again. Following the addition of a hypotonic solution (KCl; 0.075 M) onto the pellets, they were centrifuged once more and incubated at 37ºC for 30 min. Next, 2 ml of fixative (2:1; methanol: acetic acid) was added on each pellet and the samples were kept at −20ºC for 15 min. Smears were prepared on slides, air dried, treated in 1× phosphate-buffered saline (PBS) for 5 min, followed by washing in an ethyl alcohol series (70, 85 and 100%). The In Situ Cell Death Detection Kit, Fluorescein (Roche, Merck KGaA, Darmstadt, Germany) was used to prepare the stock solution, which was added onto the slides. Then slides were then covered to provide darkness, and were incubated at 37ºC for 45 min. Subsequently, they were washed three times in PBS for 1 min each, and left to dry at room temperature. Cells they were stained with DAPI, and incubated at 37ºC for 45 min covered with aluminium foil. Following the washing procedure with PBS for 1 min, samples were air dried, stained with DAPI again, and analyzed under a FITC–DAPI filter in a fluorescence microscope. Minimally, 200 sperm cells were analysed by observing at least 10 microscopic fields with a ×10 magnification. All sperm cells analyzed microscopically were chosen randomly from different parts of the slides. The sperm cells with DNA fragmentation were visualized as green, and those with no DNA fragmentation were visualized as blue.
Caspase-3 analysis
Caspase-3 expression level analysis
Caspase-3/CPP32 Colorimetric Assay Kit, Biovision was used to observe caspase-3 activation levels [optical density (OD) 405 nm]. The procedure was performed as recommended in the datasheet: 10 µl of DTT (1 M) was added to 1 ml of 2× Reaction Buffer to obtain a 10 mM solution. The sperm cells were added into the solution and counted to give a final suspension of 1 × 107. Cryptozoospermic samples were counted to give a final suspension of 1 × 106.
Results were multiplied by dilution rate (×10). Sperm counts were performed in replicates. The suspension was then re-suspended in chilling cell lysis buffer for 10 min on ice. Following centrifugation at 10,000 g for 1 min, the supernatant (cytosolic extract) was separated and kept at −80°C until analysis.
On the day of analysis, the frozen samples were thawed and protein concentration was determined as follows: samples were adjusted to have 200 µg protein in 50 µl of Cell Lysis Buffer; 50 µl of 2× Reaction Buffer (including 1 mM DTT) were added on this suspension which was taken into micro-wells. Subsequently, 5 µl of 4 mM DEVD-PNA substrate was added to each well to give a 200 µM final concentration and wells incubated at 37°C for 2 h. The absorbance values (OD) of the samples were analyzed at 405 nm by ELISA.
Caspase-3 subcellular localization analysis
Immunohistochemistry (IHC) techiques were applied to detect the localization of caspase-3 expressions. Sperm smear slides were left to air dry, then fixed with 4% paraformaldehyde. After washing with PBS three times, a 3% hydrogen peroxide–methanol mixture was applied to the slides for 20 min. Antigen retrieval was performed using citrate buffer in a microwave. Slides were air dried, then washed with PBS three times. Blocking solution of Mouse Specific HRP (ABC) Detection IHC Kit (ab93677) (ABCA, Cambridge, UK) was applied according to the manufacturer’s protocol. Primer caspase-3 antibodies were prepared as a 1/100 dilution (sc56046) (Santa Cruz Biotechnology, Inc. Texas, USA) and applied on the slides overnight in a humidified chamber as recommended by the manufacturer. The day after, slides were washed and the streptavidin peroxidase and biotinylated goat anti-polyvalent solution of the IHC kit were applied as recommended by the manufacturer. A DAB Substrate kit (ab64238) (ABCA, Cambridge, UK) was applied according to the manufacturer’s recommendations. Haematoxylin was used to measure background staining. Then the slides were mounted with bio-mount medium. At least 100 sperm cells were analyzed by light microscopy (×100) and the results were calculated as percentage expression (%).
Statistical analysis
SPSS for Windows 16.0 software package (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. The Mann–Whitney U-test was used to evaluate the differences between the groups. The results were evaluated in 95% confidence intervals and the statistical significance was defined as a P-value < 0.05.
Results
The mean age of the patients was 36.9±18 years (range: 22–58 years) and mean baseline semen volume (ml) was 4.0±2.2 ml. Semen characteristics of patients are given in Table 1. Sperm parameters (%) were investigated in 72 sperm samples prior to and after cryopreservation. Although there were no significant differences in sperm concentrations and normal morphology rate before and after cryopreservation, total motility, progressive motility and vitality rates decreased significantly after cryopreservation (P = 0.032; P = 0.040; P = 0.041 respectively).
Table 1 Comparison of semen parameters before and after cryopreservation. Values are mean±SD unless otherwise stated

NS, not significant; SD, standard deviation.
DNA fragmentation rates (%) were investigated in 72 sperm samples prior to and after cryopreservation and found to be increased significantly after cryopreservation (23.98±8.07%; 27.34±8.61% respectively) (P = 0.03). When the patients were sub-grouped as normozoospermic or non-normozoospermic, DNA fragmentation rates were found to be 23.25 and 24.71% in non-normozoospermic samples; and 26.32 and 28.36% in normozoospermic samples respectively), although the differences obtained were not statistically significant (P = 0.07 and P = 0.07, respectively) (Fig. 1).
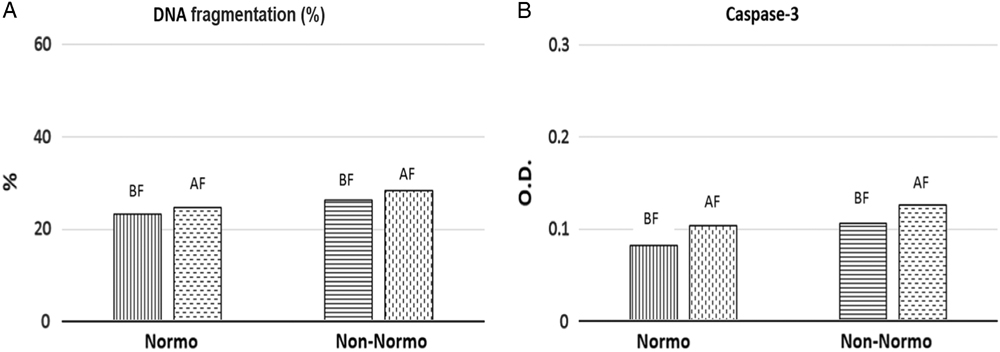
Figure 1 DNA fragmentation rates (A) and caspase-3 activation levels (optical density, OD) (B), in normozoospermic or non-normozoospermic patients. DNA fragmentation rates were given as mean (%) and caspase-3 activation levels were given as mean OD. Abbreviations: BF, before freezing; AF, after freezing; Normo, normozoospermic patients; Non-normo: non-normozoospermic patients.
Caspase-3 activation levels (OD 405 nm) were measured in 72 sperm samples before and after cryopreservation. Mean caspase-3 activation values (OD 405 nm) of the patients were calculated as 0.093±0.044 (OD 405 nm) before cryopreservation and 0.116±0.105 (OD 405 nm) after cryopreservation. Differences were not statistically significant (P = 0.06). When the patients were grouped as normozoospermic and non-normozoospermic, caspase-3 activation (OD 405 nm) values were found to be 0.082 and 0.104 (OD 405 nm) in non- normozoospermic samples; 0.106 and 0.126 ((OD 405 nm) in normozoospermic samples respectively), but differences were not statistically significant (P = 0.07 and P = 0.08, respectively) (Fig. 1).
Subcellular localization of caspase-3 protein in human sperm was also investigated before and after cryopreservation. We observed that caspase-3 was expressed in both fresh ejaculated sperm and thawed sperm after cryopreservation and was localized along the entire human spermatozoa, including the acrosome region, the midpiece, and along the tail with variable immunostaining intensity. Before cryopreservation, sperm samples showed prominent caspase-3 immunostaining at the most proximal part of the acrosome region (62% before cryopreservation and 32% after cryopreservation) and at the membrane of the head (48% before cryopreservation and 30% after cryopreservation). Whereas we observed a prominent staining at the midpiece part of the sperm (62% after cryopreservation and 32% before cryopreservation) and at the cytoplasmic droplets of abnormal sperm cells (42% after cryopreservation and 28% before cryopreservation) (Fig. 2). Interestingly, we observed weaker caspase-3 staining in two particular spermatozoa regions: the postacrosomal region of the head (12% before cryopreservation and 15% after cryopreservation) and in the tail (8% before cryopreservation and 12% after cryopreservation) both before and after cryopreservation. All of the cryopreserved sperm samples were used for intracytoplasmic sperm injection (ICSI) after thawing and resulted in an overall pregnancy rate of 32.6%, which was calculated as 39.4% of the fresh ejaculate group.
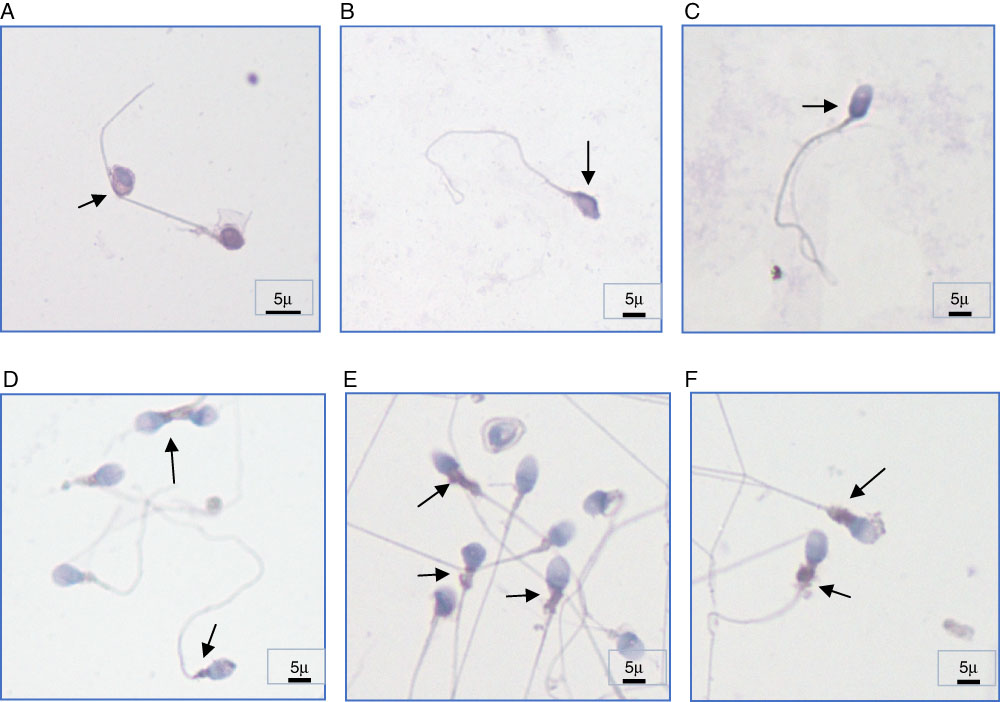
Figure 2 Subcellular localization of caspase-3 in human spermatozoa of different individuals. (A–C) Caspase-3 immunostaining regions before cryopreservation (prominent staining at the most proximal part of the acrosome region and at the membrane of the head). (D–F) Caspase-3 immunostaining regions after cryopreservation (prominent staining at the midpiece part of the sperm) ×100 magnification. Arrows indicate positive immunostaining regions. Scale bars are indicated (5 µm).
Discussion
The mechanisms underlying the negative effects of the cryopreservation process on sperm cells are not fully understood. DNA fragmentation and caspase activation are late signs of apoptosis and therefore used as markers to detect apoptosis in a cell. Caspase-3 has been implicated as an effector caspase associated with the initiation of the death cascade and is therefore an important marker of the cell’s entry point into the apoptotic signalling pathway. Caspase-3 is activated by upstream caspase-8 and caspase-9 and, as it serves as a convergence point for different signalling pathways, it is well suited as a read-out in an apoptosis assay. Several studies have reported a relationship between caspase activation, phosphatidylserine externalization and DNA fragmentation and male infertility (Weng et al., Reference Weng, Taylor, Morshedi, Schuffner, Duran, Beebe and Oehninger2002; Isachenko et al., Reference Isachenko, Isachenko, Katkov, Rahimi, Schöndorf, Mallmann, Dessole and Nawroth2004; Petyim & Choavaratana, Reference Petyim and Choavaratana2006; Grunewald et al., Reference Grunewald, Sharma, Paasch, Glander and Agarwal2009). It has also been demonstrated that the percentage of sperm cells with damaged DNA increased (>30–40%); sperm parameters (Sun et al., Reference Sun, Jurisicova and Casper1997; Zini et al., Reference Zini, Bielecki, Phang and Zenzes2001), pregnancy rates (Evenson et al., Reference Evenson, Jost, Marshall, Zinaman, Clegg, Purvis, de Angelis and Claussen1999; Spanò et al., Reference Spanò, Bonde, Hjollund, Kolstad, Cordelli and Leter2000) and fertilization rates following IVF and ICSI decreased (Lopes et al., Reference Lopes, Sun, Jurisicova, Meriano and Casper1998; Host et al., Reference Host, Lindenberg and Smidt-Jensen2000). Damaged sperm DNA led to embryos with damaged genetic material following ICSI cycles (Lopes et al., Reference Lopes, Sun, Jurisicova, Meriano and Casper1998; Ahmadi & Ng, Reference Ahmadi and Ng1999). As a result, sperm DNA fragmentation and its effects have attracted increased attention in studies, however effects of cryopreservation on the integrity of sperm DNA are controversial. Although, most studies have suggested that cryopreservation generates, as well as exacerbates, the extent of sperm DNA fragmentation in infertile patients (Hammadeh et al., Reference Hammadeh, Askari, Georg, Rosenbaum and Schmidt1999; Donnelly et al., Reference Donnelly, McClure and Lewis2001; de Paula et al., Reference de Paula, Bertolla, Spaine, Cunha, Schor and Cedenho2006; Thomson et al., Reference Thomson, Fleming, Aitken, De Iuliis, Zieschang and Clark2009a,Reference Thomson, Fleming, Barone, Zieschang and Clarkb,Reference Thomson, Fleming, Schulke, Barone, Zieschang and Clarkc), others have reported no effect of cryopreservation on DNA integrity (Duru et al., Reference Duru, Morshedi, Schuffner and Oehninger2001; Isachenko et al., Reference Isachenko, Isachenko, Katkov, Rahimi, Schöndorf, Mallmann, Dessole and Nawroth2004). Spanò and colleagues demonstrated that sperm DNA integrity was negatively affected by sperm cryopreservation procedures in 19 cases in which the SCSA test was used to analyze DNA content (Spanò et al., Reference Spanò, Cordelli, Leter, Lombardo, Lenzi and Gandini1999). Similar results were obtained by de Paula and colleagues in their study of 77 cases, in which the TUNEL assay was used (de Paula et al., Reference de Paula, Bertolla, Spaine, Cunha, Schor and Cedenho2006). In contrast, Isachenko and colleagues demonstrated using two different freezing methods (vitrification and slow freezing) that there was no effect of cryopreservation on DNA integrity. In the present study, we observed a significant increase in sperm DNA fragmentation rates in all of the patient samples after cryopreservation, regardless of the quality of the semen, when the slow freezing and TUNEL methods were used. Thia diversity may be due to the different cryopreservation techniques, DNA fragmentation assessment methods, and different sample sizes. The study of Petyim & Choavaratana (Reference Petyim and Choavaratana2006) justified this conclusion when they reported different DNA fragmentation rates in their study comparing two different cryopreservation. When we sub-grouped the patients as normozoospermic or non-normozoospermic, we observed that DNA fragmentation rates were similar between these groups with different sperm parameters before and after cryopreservation as it is the cryopreservation process itself that causes DNA fragmentation, not the quality of the semen sample.
The second parameter analyzed was caspase-3 activation status. Cryopreservation has been shown to increase the activation of caspases in humans in several studies (Paasch et al., 2004; Wündrich et al., Reference Wündrich, Paasch, Leicht and Glander2006). It has been suggested that DNA fragmentation was partially a consequence of caspase-3 activation (Weng et al., Reference Weng, Taylor, Morshedi, Schuffner, Duran, Beebe and Oehninger2002; Grunewald et al., Reference Grunewald, Sharma, Paasch, Glander and Agarwal2009). However, Paasch et al. (2004) in their study of 84 samples observed no increase in sperm DNA fragmentation levels, although activation of caspases 3, 8 and 9 was observed following cryopreservation. We showed that DNA fragmentation was increased in 252 samples but caspase-3 levels were not affected after cryopreservation. We observed that caspase-3 activation levels were similar before and after cryopreservation in 72 individuals. When the patients were grouped as normozoospermic or non-normozoospermic, we found no significant difference between these subgroups. These results indicated that caspase-3 activation was neither affected by the cryopreservation process, nor by the quality of the sample. The only difference observed for caspase-3 was localization of this protein’s expression before and after cryopreservation. Although caspase-3 is expressed in both fresh ejaculated sperm and thawed sperm after cryopreservation and is localized along the entire human spermatozoa, at the acrosome, midpiece and along the tail with variable staining intensity. Sperm samples showed prominent immunostaining at the most proximal parts of the acrosome region and at the membrane of the head before cryopreservation but prominent staining at the midpiece part of the sperm and in the cytoplasmic droplets of abnormal sperm cells after cryopreservation. Caspase-3 immunostaining in the postacrosomal region of the head and in the sperm tail was weaker in both samples before and after cryopreservation.
According to our results, it may be concluded that the cryopreservation procedure has a negative effect on these sperm parameters, but when apoptotic signs are considered, there was no significant difference in sperm DNA fragmentation levels and caspase-3 activation levels. The values obtained for both apoptotic markers did not differ between the normozoospermic and non-normozoospermic subgroups, indicating that sperm sample quality had no effect on these apoptotic markers during cryopreservation, instead the cryopreservation procedure itself affected DNA fragmentation levels negatively. The differences obtained for caspase-3 localization may be the result of different signalling pathways induced as a consequence of physical stress driven by the cryopreservation process. Caspase-3 protein levels were higher in the midpiece region of the sperm after cryopreservation. Because mitochondria are mostly located in the midpiece region of the sperm cells these results may reflect the increasing activity of mitochondria to overcome stress produced by cryodamage and trigger the mitochondria-mediated intrinsic apoptotic pathway.
In conclusion, the results obtained in the present study may help to explain the causes of cryodamage observed after cryopreservation and to help investigators to develop strategies to avoid or minimize the negative effects observed. Furthermore, the results may provide valuable information when comparing different methods of sperm cryopreservation, DNA fragmentation and different sperm qualities. Further studies that observe more outcome apoptotic markers and with larger sample sizes should be conducted to analyse the effects of cryopreservation on different sperm subpopulations.
Financial support
This research received no specific grant from any funding agency, or commercial or not-for-profit sectors.
Conflicts of interest
There are no conflicts of interest.
Ethical standards
All the patients were informed and gave written consent. The ethical committee of Istanbul Medipol University approved the study on 13 November 2017 and the study was given ethical number 10840098-604.01.01-E.42838.



