

This article seeks to identify some important neurological disorders that might present with psychiatric comorbidity. These can occur as comorbidity in children with an established diagnosis such as Tourette syndrome, but they also arise in children for whom no coexisting diagnosis/syndrome has yet been considered and in children with learning disability. Alternatively, children and adolescents with a neurodegenerative disease may present initially with a psychiatric/behavioural disorder, e.g. the early affective disorder associated with new variant Creutzfeld–Jakob disease (the human variant of bovine spongiform encephalopathy (BSE)). Thus, the paediatric psychiatric assessment offers a wide-ranging diagnostic opportunity.
Information concerning developmental history and the observation of play that routinely form part of a child psychiatric assessment is also relevant to the paediatric neurological assessment. Some additional interpretation of these observations may be needed: for example handedness and the quality of motor movements during play, as well as its style and content, are important. As in child psychiatry, the child neurologist learns more during observation of play or spontaneous activity than from a formal neurological examination.
Dysmorphic features
The assessment of dysmorphic features is subjective and should take place after both parents have been seen. This will avoid offence to whichever parent the child closely resembles and may avoid a prolonged search for a syndromic diagnosis. Conversely, the child may have a diagnosis in common with a parent, inherited in an autosomal dominant fashion. Table 1 contains examples of syndromic diagnoses associated with dysmorphism, along with other phenotypic features that may precipitate the involvement of child and adolescent mental health services (CAMHS).
Table 1 Syndromic diagnoses associated with dysmorphism

| Syndrome | Physical and neuropsychiatric characteristics |
|---|---|
| Fragile-X syndrome (X-linked) | Physical: macrocephaly and testicular enlargement in early childhood, prognathism after puberty, large ears |
| Neuropsychiatric: mild-to-profound learning disability, hyperactive behaviour, emotional instability, autistic features (60%), cluttered or repetitive speech | |
| Cornelia de Lange syndrome (sporadic and autosomal dominant) | Physical: small stature, brachycephaly, bushy eyebrows with synophrys and long curled eyelashes, low posterior hairline |
| Neuropsychiatric: moderate-to-severe learning difficulties, autistic and self-destructive behaviour, rejection of physical contact, enjoyment of rapid movement but otherwise emotionless | |
| Prader–Willi syndrome (sporadic) | Physical: obesity, strabismus, small stature along with small hands and feet, small genitalia and cryptorchidism |
| Neuropsychiatric: moderate-to-severe learning difficulties, stubborness, rages, verbal perseverance on favourite topics, overwhelming compulsion to eat | |
| Angelman syndrome (sporadic) | Physical: maxillary hypoplasia, deep-set eyes, large mouth with tongue protrusion prognathia, widely spaced teeth, characteristic arm posture with elbows and wrists in flexion, ataxic gait |
| Neuropsychiatric: severe learning difficulties with little or no speech, paroxysms of inappropriate laughter, seizures, little need for sleep |
Neurocutaneous stigmata
Certain conditions are associated with particular cutaneous manifestations and may present to paediatric services, paediatric neurologists or psychiatrists because of associated neurological, behavioural or developmental problems. Therefore the skin should always be examined to identify depigmented areas; White Caucasians should ideally be examined using a Wood's light as well as the naked eye. Table 2 contains examples of conditions with neurocutaneous stigmata which may present to CAMHS. Neurofibromatosis type 1 (NF1) and tuberous sclerosis are discussed further, since they are common conditions with significant neuropsychiatric difficulties.
Table 2 Conditions with neurocutaneous stigmata

| Syndrome | Physical and neuropsychiatric characteristics |
|---|---|
| Neurofibromatosis | Physical: café-au-lait spots after 1 year of age, axillary or inguinal freckling by 3 years, cutaneous and plexiform neurofibromas |
| Neuropsychiatric: seizures, headaches, speech and language deficits, mild-to-moderate learning difficulties, immature coordination, hyperactivity, anxiety, depression | |
| Tuberous sclerosis | Physical: depigmented macules, ash-leaf macules, shagreen patches, adenoma sebaceum from 5 years of age, periungual fibromas, retinal phakomas |
| Neuropsychiatric: vast range of cognitive ability from profound disability to superior performance, seizures (including infantile spasms), aggression, autistic features in up to 50% of cases and in all with infantile spasms, stereotypies, severe sleep disturbance, uncontrollable screaming, bouts of destructive behaviour | |
| Sturge–Weber syndrome | Physical: pink/purple (port-wine coloured) non-elevated haemangiomas most commonly in a trigeminal facial distribution, occasionally bilateral, flame-like naevi may be present over the trunk |
| Neuropsychiatric: seizures, learning difficulties which may be progressive, hemiplegia, hemianopia |
Neurofibromatosis type 1
Neurofibromatosis type 1 has an incidence of 1 in 2500–4000 live births across all ethnic and racial groups. It is inherited in an autosomal dominant manner, with 98–100% penetrance, and similar neurocutaneous features may be found during examination of the parents’ skin. However, the new-mutation rate is high and accounts for about 30% of cases, so parents may not be affected (Reference AicardiAicardi, 1998). The appearance of the skin varies from minor coffee-coloured macules and axillary freckling to significantly disfiguring neurofibromas. Macrocephaly may be observed in up to 43% of those with this condition and short stature in about 27% (Reference NorthNorth, 1993).
Neurofibromas are benign tumours or hamartomas which may be located intracutaneously as violaceous nodules, subcutaneously along the nerve sheath, or in the iris as Lisch nodules. The criteria for the diagnosis of neurofibromatosis are shown in Box 1 (Reference AicardiAicardi, 1998). Clinicians should be alert to any reported change in vision in these patients, since optic nerve gliomas affect some 15–20% (Reference AicardiAicardi, 1998). However, lesions at the chiasm produce a bi-temporal hemianopia, which frequently goes unnoticed by the patient, making it mandatory to undertake a clinical assessment of visual fields.
Box 1 Criteria for the diagnosis of neurofibromatosis type 1 (after Reference AicardiAicardi, 1998)
The diagnosis may be made in the presence of two or more of the following criteria:
Six or more café-au-lait spots >5 mm in diameter in pre-pubertal patients and >15 mm in post-pubertal patients
Two or more neurofibromas (intracutaneous or subcutaneous) or one plexiform neurofibroma
Freckling in the axillary or inguinal regions
Optic glioma
Two or more iris hamartomas (Lisch nodules)
Typical osseous lesion such as sphenoid dysplasia and tibial pseudarthrosis
One or more first-degree relatives with neurofibromatosis type 1 (NF-1)
Epilepsy is seen in up to 7% of children with NF1, with all seizure types represented. Although the presence of severe learning disability is rare, moderate learning disability occurs in 10% of cases and the IQ of patients with NF1 is, on average, lower than that of unaffected siblings. Specific learning difficulties affect 40–60% of patients and include visuospatial deficits, perceptual organisation difficulties, poor coordination, poor spatial and verbal memory, reading and writing difficulties and problems with numeracy (Reference Udwin, Dennis, O'Brien and YuleUdwin & Dennis, 1995). Up to 50% of children with NF1 have speech and language difficulties (Reference Udwin, Dennis, O'Brien and YuleUdwin & Dennis, 1995), including excessive nasality, breathiness and slow acquisition of vocabulary, although overall verbal abilities tend to be higher than non-verbal ones.
Attention difficulties in the absence of hyperactivity have been described (Reference AicardiAicardi, 1998) and, interestingly, distractibility and poor concentration seem more common in children with greater ability. Unhappiness and victimisation by bullying are frequently reported, as are anxiety, poor self-confidence and social withdrawal, and an excess of psychiatric morbidity such as anxiety and depression.
Tuberous sclerosis
The prevalence of tuberous sclerosis is reported to be of the order of 1 in 7000 of the population. It is an autosomal dominant condition with a very high rate of spontaneous mutations (58–68%). Hypomelanotic macules, commonly in an ash-leaf shape, are the most frequent cutaneous manifestation of tuberous sclerosis and are best seen using a Wood's light. Shagreen patches are yellow/brown elevated layers of epidermis with a granular surface seen in the lumbar region after the age of 2 years. Periungual fibromas, although pathognomonic, rarely appear before puberty. The best-known cutaneous manifestation of tuberous sclerosis is adenoma sebaceum, the papular, acneiform rash that appears in a butterfly distribution over the face between 3 and 15 years of age. Retinal astrocytomas or phakomas are round/oval lesions involving the optic nerve and retina which, when calcified, resemble mulberries.
The characteristic features of the syndrome are shown in Box 2 (Reference AicardiAicardi, 1998). It is a multi-system disorder with effects in the brain, heart, skin, kidneys and lungs. The most characteristic findings in those with tuberous sclerosis relate to the brain. They are cortical tubers, subependymal nodules and the subependymal giant cell astrocytomas that result in hydrocephalus. Seizures are the most common presenting complaint, occurring in 60% of individuals (Reference Webb, Fryer and OsborneWebb et al, 1991), with the most commonly described being infantile spasms. However, the full range of seizures may be seen, including complex partial, tonic and atonic attacks.
Box 2 Diagnostic criteria for tuberous sclerosis (after Reference AicardiAicardi, 1998)
Pathognomonic features
Cortical tuber
Subependymal glial nodules
Retinal phakomas
Adenoma sebaceum
Periungual fibroma
Fibrous plaque of forehead/scalp
Multiple renal angiomyolipomas
Suggestive features
Immediate relative with tuberous sclerosis
Hypomelanotic macules
Shagreen patches
Cardiac rhabdomyomas
Gingival fibroma
Multiple renal tumours
Renal cysts
Infantile spasms
Wedge-shaped areas of cortical/subcortical calcification
Multiple subcortical hypodense areas on computed tomography (or high T2 signal on magnetic resonance imaging)
Peripapillary retinal hamartoma
Pulmonary lymphangiomyomatosis
Fewer than 50% of those affected are thought to have a learning disability (Reference Webb, Fryer and OsborneWebb et al, 1991), although when present, this may be profound (IQ < 20). Cognitive performance seems to be adversely associated with epilepsy, particularly with seizure onset below the age of 2 years. A prevalence study in the west of Scotland (Reference Hunt and ShepherdHunt & Shepherd, 1993) showed that 43% of children with tuberous sclerosis fulfilled DSM–III–R (American Psychiatric Association, 1987) criteria for autism or pervasive developmental disorder. Reference Gillberg, Gillberg and AhlsenGillberg et al(1994) reported autism in 61% of those below 20 years of age with tuberous sclerosis and found many with attention-deficit hyperactivity disorder. Sleep disorder is common and particularly trying for the family. It is associated with the presence and severity of epilepsy. The behaviour seen in children with tuberous sclerosis means that they can rarely be left unsupervised as they may exhibit destructive behaviour. They seem impervious to verbal suggestion and appear to be in their own world (Reference Udwin, Dennis, O'Brien and YuleUdwin & Dennis, 1995). Attempts to redirect their activity provoke aggression and, in severe cases, the family may view trips out of the house as being too difficult to contemplate.
Occipitofrontal circumference, weight and height
As in any other paediatric examination, it is mandatory to plot weight and height on percentile charts when undertaking a neurological examination. As well as drawing attention to the possibility of eating disorders or depression, these measurements may also identify abnormal growth such as the short stature seen in a number of syndromes, including Noonan's syndrome and Cornelia de Lange syndrome, or the tendency to early overgrowth seen in fragile-X syndrome or Sotos syndrome. Obesity may also be a clue to an underlying syndromic diagnosis such as Prader–Willi syndrome or Cohen syndrome.
Plotting the occipitofrontal circumference (OFC) is part of the paediatric neurological examination at all ages. Head circumference measurements from both parents may be relevant, since familial macrocephaly does occur, affects more boys than girls and is observed more commonly in fathers than in mothers (Reference Day and SchuttDay & Schutt, 1979). To encourage children's cooperation it is useful to pretend to be measuring the family for hats; with babies or toddlers leave the assessment until the end of the examination. Special charts are available for OFC (some general growth charts include this only up to 1 year of age). Sequential measurements are more useful than one-off measurements in the interpretation of head size, as head circumference measurements that progressively cross percentiles are more concerning than is a large or small head from birth which is growing along the same percentile. Table 3 shows some causes of macrocephaly and microcephaly and their particular neuropsychiatric associations.
Table 3 Conditions with variation in head size
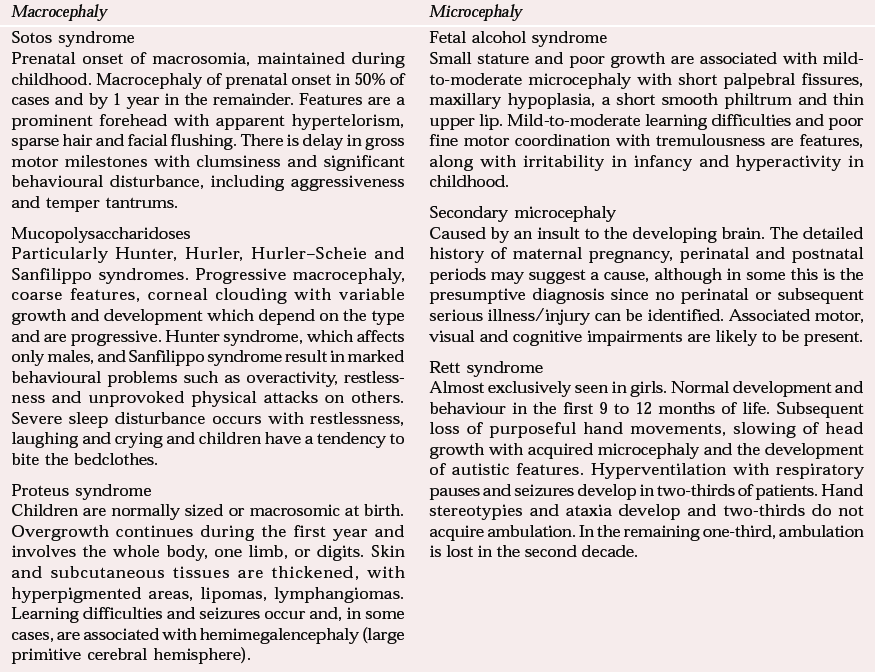
| Macrocephaly | Microcephaly |
|---|---|
| Sotos syndrome | Fetal alcohol syndrome |
| Prenatal onset of macrosomia, maintained during childhood. Macrocephaly of prenatal onset in 50% of cases and by 1 year in the remainder. Features are a prominent forehead with apparent hypertelorism, sparse hair and facial flushing. There is delay in gross motor milestones with clumsiness and significant behavioural disturbance, including aggressiveness and temper tantrums. | Small stature and poor growth are associated with mild-to-moderate microcephaly with short palpebral fissures, maxillary hypoplasia, a short smooth philtrum and thin upper lip. Mild-to-moderate learning difficulties and poor fine motor coordination with tremulousness are features, along with irritability in infancy and hyperactivity in childhood. |
| Mucopolysaccharidoses | Secondary microcephaly |
| Particularly Hunter, Hurler, Hurler–Scheie and Sanfilippo syndromes. Progressive macrocephaly, coarse features, corneal clouding with variable growth and development which depend on the type and are progressive. Hunter syndrome, which affects only males, and Sanfilippo syndrome result in marked behavioural problems such as overactivity, restlessness and unprovoked physical attacks on others. Severe sleep disturbance occurs with restlessness, laughing and crying and children have a tendency to bite the bedclothes. | Caused by an insult to the developing brain. The detailed history of maternal pregnancy, perinatal and postnatal periods may suggest a cause, although in some this is the presumptive diagnosis since no perinatal or subsequent serious illness/injury can be identified. Associated motor, visual and cognitive impairments are likely to be present. |
| Proteus syndrome | Rett syndrome |
| Children are normally sized or macrosomic at birth. Overgrowth continues during the first year and involves the whole body, one limb, or digits. Skin and subcutaneous tissues are thickened, with hyperpigmented areas, lipomas, lymphangiomas. Learning difficulties and seizures occur and, in some cases, are associated with hemimegalencephaly (large primitive cerebral hemisphere). | Almost exclusively seen in girls. Normal development and behaviour in the first 9 to 12 months of life. Subsequent loss of purposeful hand movements, slowing of head growth with acquired microcephaly and the development of autistic features. Hyperventilation with respiratory pauses and seizures develop in two-thirds of patients. Hand stereotypies and ataxia develop and two-thirds do not acquire ambulation. In the remaining one-third, ambulation is lost in the second decade. |
In terms of behavioural phenotype, Sotos syndrome is one of the most interesting. Children with this syndrome are large at birth, with advanced bone age and accelerated growth, particularly in the first 5 years. Growth continues above the 97th percentile for height and head circumference but later falls to within the normal range. Early motor and language milestones are delayed and children experience fine motor and coordination difficulties. Cognitive abilities are varied and range from severe impairment to average intelligence, but the profile is uneven, with a higher level of difficulty in verbal processing and short-term memory. Behavioural problems are usually more evident at home than at school and consist of aggressive behaviour and temper tantrums. Children with Sotos syndrome exhibit social and emotional immaturity and have difficulties with concentration and hyperactivity that appear exaggerated because these individuals do not perform at the level expected for their size.
Rett syndrome (Table 3) deserves special mention as a cause of acquired microcephaly. The gene responsible for the disorder is thought to be located on the X chromosome. As the children (who are mostly girls) become older, physical disabilities increase, with hypertonia, spasticity and progressive scoliosis. Psychological/behavioural difficulties include anxiety, panic and screaming (Reference Udwin, Dennis, O'Brien and YuleUdwin & Dennis, 1995), which can be provoked by sudden noises or changes of routine. Self-injury is a prominent feature and is associated with hyperventilation and general distress. Sleep disturbance is very common and includes delayed onset of sleep and nocturnal wakening with screaming and laughing.
Cranial nerves
Eyes
The eyes can yield many clues for potential neurological diagnoses and, as far as possible, every patient should be examined. Table 4 lists some ophthalmological features in patients who might present to CAMHS, with associated diagnoses. The most obvious ophthalmological observation on first seeing the patient is that he or she is wearing thick spectacles. Bardet–Biedl syndrome, once referred to as Laurence–Moon–Biedl syndrome, has a variety of ocular features, including retinal dystrophy, astigmatism, glaucoma and retinitis pigmentosa. Other features include post-axial polydactyly, hypogonadism and obesity. Moderate learning difficulties are also seen, along with a shallow affect and inappropriate mannerisms (Reference JonesJones, 1997).
Table 4 Diagnostic clues from examination of the eyes

| Acuity | Myopia is seen in Bardet–Biedl syndrome and Rubenstein–Taybi syndrome |
| Fields | Hemianopia or partial-field defects associated with hemiplegia, bitemporal hemianopia associated with lesions at the chiasm |
| Cornea/lens | Lisch nodules of neurofibromatosis, cataract in Lowe's syndrome, lens dislocation in homocystinuria and Marfan syndrome, telangiectasia in ataxia-telangiectasia aged >3 years, Kayser–Fleischer rings in Wilson's disease aged >8 years |
| Fundi | |
| Optic atrophy | Leukodystrophy, gangliosidoses |
| Papilloedema | Evolution of hydrocephalus in neurofibromatosis or tuberous sclerosis |
| Phakomas | Tuberous sclerosis |
| Cherry-red spots | Gangliosidoses, sialidosis |
| Pigmentary changes | Mitochondrial disease, juvenile neuronal ceroid lipofuscinosis (Batten disease), Usher syndrome |
| Colobomas | Colomba, heart disease, atresia of the choanae, retarded growth and devlopment, genital abnormlaities and ear abnormalities, including deafness (the CHARGE association), Cat-eye syndrome |
| Eye movements and nystagmus | Juvenile Gaucher disease, ataxia telangiectasia, Niemann–Pick disease type C, spinocerebellar degeneration, Huntington's chorea. |
It can be difficult to assess visual fields in young children or in those whose development has been delayed, but this should be attempted, especially in the presence of a motor deficit where a coexisting visual-field deficit may explain reduced visual awareness of material presented to that side. A useful method of assessment, if examining alone, is to hold two fingers out to either side within the child's lateral visual fields and midway between yourself and the child. Then call the child's name so that he or she looks at you and ask which fingers are moving. Move the fingers on one side and the child will either look or point at them. Call to the child again to bring attention back to your face and repeat the exercise for the other side and for the vertical plane. If an assistant is available a lighted torch can be introduced into the child's visual field from behind by one examiner while the other interests the child in looking forwards. The child will turn to the object when it has been sighted.
Table 4 contains examples of neurodegenerative disorders that might present later to CAMHS with a change in personality and/or behaviour, possibly associated with reduced academic performance and accompanied by evidence of visual failure. Deterioration in vision accompanied by personality change, intellectual deterioration or motor difficulties is very ominous and requires urgent further investigation. In these circumstances, fundal examination and the examination of eye movements may reveal clues as to the aetiology of the disorder.
In order to examine the fundi it is useful to ask a parent or carer to stand in front of the child and to ask the child to tell you if the adult is making faces behind your back. Most children will avidly concentrate on their parent/carer's face, allowing a view of the fundi to be obtained. Older children will usually voluntarily fix on a toy or picture but younger children or toddlers may need their interest to be captured by holding up a toy such as a teddy bear which is made to wave occasionally. Avoid using a toy that is too exciting, as the child will become upset if not allowed closer access to it and the opportunity will be lost. Pale optic discs indicative of optic atrophy, associated with evidence of visual failure, are a feature of neurodegenerative disorders such as leukodystrophy and other lysosomal enzyme disorders. These disorders are progressive and motor and mental deterioration occur before premature death. Cherry-red retinal spots may disclose the presence of GM1 gangliosidosis or, rarely, the ‘cherry-red spot-myoclonus syndrome’ (type 1 sialidosis). Pigmentary changes in the retina may be seen with mitochondrial disease.
Other significant neurological disorders are revealed by the presence of abnormal eye movements, which can be tested by moving a developmentally appropriate visual lure. Supranuclear ophthalmoplegia refers to a disturbance in the mechanisms controlling voluntary conjugate gaze, the cause of which lies above the level of those cranial nerve nuclei that control eye movements. Diffuse cerebral disease of any cause may affect both saccadic and pursuit eye movements. In affected children, voluntary side-to-side eye movements (saccades) are hypometric and slow, necessitating the use of a head thrust or forced blinking in order to relocate the gaze on a target. Pursuit movements in these children are small and jerky on following a target. The vestibulo-ocular brainstem reflex, which is independent of voluntary mechanisms, remains unaffected. Conditions producing a supranuclear ophthalmoplegia include juvenile Gaucher disease (supranuclear horizontal ophthalmoplegia), the adult form of GM2 gangliosidosis and type C Niemann–Pick disease (supranuclear vertical ophthalmoplegia). All of these autosomal recessive disorders are progressive, with variable rates of concurrent or subsequent deterioration in motor and intellectual function.
The disorder most familiar to a paediatric neurologist is that of ataxia-telangiectasia, a disorder with autosomal recessive inheritance. Clinical features include progressive cerebellar ataxia with choreo-athetosis in 25% of patients, progressive oculocutaneous telangiectasias, immunodeficiency and a high incidence of malignancy. Although the ataxia and apraxic eye movements are present after the age of 1 year, these might not have been recognised and the child may not be correctly diagnosed with the disorder until later in childhood, when up to one-third show mild learning difficulties (Reference AicardiAicardi, 1998). In most cases, dilated conjunctival vessels in the angles of the eye are noted after the age of 3 years and these spread medially. The course is relentless and the prognosis poor: children are wheelchair-bound by 10 to 15 years of age.
Nystagmus is an involuntary, rhythmical conjugate oscillatory movement of the eyes, it may occur in any plane and may be jerky or pendular, or both. It is important to differentiate true nystagmus from the roving eye movements of the blind child. The most common type is gaze-evoked nystagmus which is jerky and is produced by asking the patient to deviate his or her eyes to either side. This usually reflects dysfunction in the posterior fossa. Impairment of the vestibular tract anywhere from nucleus to end organ can also produce jerky nystagmus. Congenital nystagmus will have been present since shortly after birth and a history of this is usually readily obtainable from a parent or carer. It is usually seen as large-amplitude, pendular, horizontal nystagmus in a child who is otherwise neurologically normal, and it may be present in a parent.
Nystagmus is not a feature of cerebral palsy, including ataxic cerebral palsy, so it should not be accepted as such without further investigation. In boys, the presence of ataxia with nystagmus and variably progressive pyramidal weakness can indicate Pelizaeus–Merzbacher disease, an X-linked leukodystrophy, the first manifestation of which is rotatory nystagmus presenting in the first 3 months of life and variably beyond this time. Unfortunately, this is also a relentlessly progressive disorder and, as in all these disorders, the correct diagnosis is important for the genetic counselling of the parents and siblings and for discussion of the availability of prenatal diagnosis in subsequent pregnancies.
Remaining cranial nerves
Observations should be made of facial symmetry. In upper motor neuron facial weakness, movement of the eyelid, eyebrow and forehead is spared. In congenital hemiplegia, facial weakness is either absent or mild, in contrast with the more marked facial weakness in acquired hemiplegia (Reference Brown, Van Rensburg and WalshBrown et al, 1987).
Oromotor movements should be assessed in any child with a history of drooling and feeding problems along with impaired speech and in whom these problems are more severe than any motor impairments. Worster–Drought syndrome consists of congenital upper motor neuron bulbar palsy with mild pyramidal tetraplegia. Affected children dribble (86%), need a modified diet (80%) and use augmentative communication (80%) (Reference Clark, Carr and ReillyClark et al, 2000). Most affected children have significant comorbidity: 81% have learning difficulty, 41% neuropsychiatric problems and 28% epilepsy. The disease is a static disorder that falls within the spectrum of cerebral palsy, often associated with bilateral perisylvian cortical dysplasia.
Problems with posture and movement
Examples of disorders that manifest progressive disturbance of posture and movement in addition to cognitive and behavioural decline are given in Table 5. A few of the more common ones are discussed below.
Table 5 Abnormal patterns of movement in progressive disorders
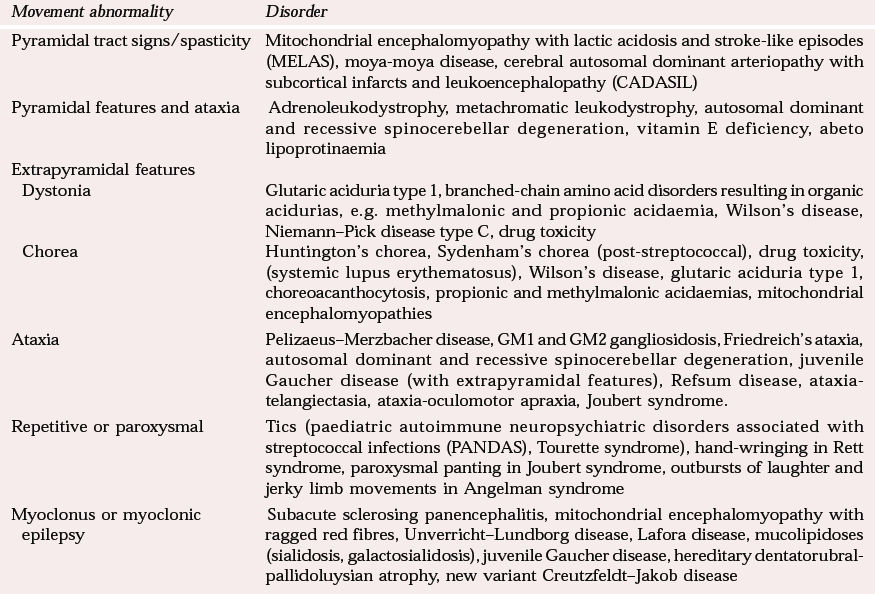
| Movement abnormality | Disorder |
|---|---|
| Pyramidal tract signs/spasticity | Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), moya-moya disease, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) |
| Pyramidal features and ataxia | Adrenoleukodystrophy, metachromatic leukodystrophy, autosomal dominant and recessive spinocerebellar degeneration, vitamin E deficiency, abeto lipoprotinaemia |
| Extrapyramidal features | |
| Dystonia | Glutaric aciduria type 1, branched-chain amino acid disorders resulting in organic acidurias, e.g. methylmalonic and propionic acidaemia, Wilson's disease, Niemann–Pick disease type C, drug toxicity |
| Chorea | Huntington's chorea, Sydenham's chorea (post-streptococcal), drug toxicity, (systemic lupus erythematosus), Wilson's disease, glutaric aciduria type 1, choreoacanthocytosis, propionic and methylmalonic acidaemias, mitochondrial encephalomyopathies |
| Ataxia | Pelizaeus–Merzbacher disease, GM1 and GM2 gangliosidosis, Friedreich's ataxia, autosomal dominant and recessive spinocerebellar degeneration, juvenile Gaucher disease (with extrapyramidal features), Refsum disease, ataxia-telangiectasia, ataxia-oculomotor apraxia, Joubert syndrome. |
| Repetitive or paroxysmal | Tics (paediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS), Tourette syndrome), hand-wringing in Rett syndrome, paroxysmal panting in Joubert syndrome, outbursts of laughter and jerky limb movements in Angelman syndrome |
| Myoclonus or myoclonic epilepsy | Subacute sclerosing panencephalitis, mitochondrial encephalomyopathy with ragged red fibres, Unverricht–Lundborg disease, Lafora disease, mucolipidoses (sialidosis, galactosialidosis), juvenile Gaucher disease, hereditary dentatorubral-pallidoluysian atrophy, new variant Creutzfeldt–Jakob disease |
To elicit evidence of dysfunction in posture and movement it can be helpful to engage the child in a game, for example with a ball. Gait, power and coordination can be observed as the ball is caught, thrown or kicked and the exercise will also put the child at ease. Toe-walking may suggest lower-limb muscle weakness with or without spasticity. Impaired walking on the heels indicates foot dorsiflexion weakness, whereas difficulty hopping, standing from a squat or climbing stairs are suggestive of weakness in the pelvic girdle. Such weaknesses are seen in muscular disorders, some of which are associated with learning impairment and behavioural disturbance (e.g. Duchenne and Becker muscular dystrophy). Children with delay or disorder of developmental coordination will also find it difficult to walk on their heels or the outsides of their feet (Fogg test) and will show excessive associated movements and postures of their arms and sometimes their mouths.
In younger children, the examination of reflexes is most easily performed with the child sitting on the parent/carer's lap resting his or her legs in the examiner's lap. The parent/carer may have to restrain one arm while reflexes are elicited in the other and giving the child a toy to play with helps during examination of lower-limb reflexes. Plantar responses are best left until last and should be performed quickly in series, since the child may find them unpleasant.
Pyramidal signs
The presence of pyramidal signs refers to the finding of increased tone, weakness which is more obvious in the extensor muscle groups of the upper limb and in the flexor muscle groups of the lower limb, and brisk tendon reflexes with an extensor plantar response. This indicates dysfunction of the corticospinal (pyramidal) tracts anywhere from the cerebral cortex to the spinal cord. Cerebral palsy is ‘an umbrella term covering a group of non-progressive, but often changing, motor impairment syndromes secondary to lesions or anomalies of the brain arising in the early stages of development’ (Reference Badawi, Watson and PettersonBadawi et al, 1998). Thus, no progression of the motor impairment should be seen if the diagnosis of cerebral palsy is correct and, as a general rule, the impairment should not evolve to include ataxia, disordered eye movements or significant extrapyramidal features.
Congenital hemiplegia is associated with significant learning and behavioural problems, which should be anticipated in order to meet the child's needs in education and within the family. Children with such impairments have significant peer difficulties, which can be predicted from lower IQ and problems such as hyperactivity reported by teachers (Reference Yude and GoodmanYude & Goodman, 1999). Psychiatric problems are common and persistent, and in the school years hyperactivity again seems predictive of continuing psychiatric morbidity. Evidence suggests that of those requiring psychiatric help, 70% will still need this help in 4 years’ time (Reference GoodmanGoodman, 1998).
Ataxia
Ataxia is defined as incoordinated or inaccurate movement which is not due to paresis, alteration in tone, loss of postural sense or the intrusion of involuntary movements (Reference PattenPatten, 1998). However, ataxia may coexist with any of the abovementioned findings in a mixed rather than pure disorder. Evolution of ataxia alongside pyramidal-tract signs associated with even the mildest cognitive or behavioural decline is indicative of a progressive spinocerebellar degeneration. In boys, this should prompt a search for X-linked adrenoleukodystrophy, a disorder of the metabolism of very-long-chain fatty acids, which presents between the ages of 4 and 8 years with gait disturbance and subtle cognitive decline. The spasticity and dementia progress and death results within a few months to several years (Reference AicardiAicardi, 1998). Metachromatic and Krabbe leukodystrophies can present in a similar way. Juvenile- and adult-onset metachromatic leukodystrophy, in particular, may present with regressive behaviour and psychiatric disease.
Ataxic cerebral palsy is a non-progressive ataxia, mainly affecting the trunk and walking balance. It is a heterogenous condition with variable aetiology. Prenatal aetiology is the most common but inherited forms of non-progressive ataxia and those associated with structural malformations of the cerebellum are also described (Reference AicardiAicardi, 1998).
Involuntary movements
Involuntary movements include tics, stereotypies, chorea, dystonia and myoclonus. The evolution of any involuntary movement disorder requires close observation and further investigation.
Tics
A tic is a sudden, rapid, recurrent, non-rhythmic stereotypic motor movement or vocalisation. Childhood tic disorder may be inherited in an autosomal dominant manner and may remit spontaneously. Tourette syndrome is a disorder in which the occurrence both of motor tics and of one or more vocal tics waxes and wanes. There is significant comorbidity with obsessive–compulsive disorder, attention-deficit and hyperactivity disorder, self-injurious behaviour, and anxiety and depression (Reference RobertsonRobertson, 2000).
Stereotypies
Stereotypies may be seen in learning disability but they are particularly associated with autism. Repetitive behaviours such as hand flapping, knuckle-biting and waving of objects may be seen in fragile-X syndrome, and repetitive hand-wringing/clapping in girls is typical of Rett syndrome.
Chorea and dystonia
Chorea and dystonia may coexist or may be the presenting feature of the same underlying disorder in different individuals which results in dysfunction of the extrapyramidal system or basal ganglia. Chorea is characterised by continuous, randomly distributed and irregularly timed muscle jerks (Reference MarsdenMarsden, 1998), which intrude into purposeful movement and speech. Dystonia consists of sustained irregular muscle spasms that last longer than chorea or myoclonus and distort the body or limb into twisted postures (Reference MarsdenMarsden, 1998). Some causes of chorea and dystonia are listed in Table 5. Two of these conditions warrant closer examination because they are relatively common.
The first is Huntington's chorea, a dominantly inherited disease, of paternal origin in 70–90% of cases. The disease most often presents in adulthood but childhood presentation is well described. Isolated mental deterioration can precede the onset of extrapyramidal features. When evident, rigidity is the dominant extrapyramidal feature in children (Reference Osborne, Munson and BurmanOsborne et al, 1982). Seizures occur in 50% of childhood cases and dysarthria is an early and prominent sign.
The second condition worthy of discussion is Wilson's disease, an autosomal recessive disorder that affects the basal ganglia and causes cirrhosis of the liver. Identification of this disorder is important since it is treatable. Thirty per cent of affected children will present with neurological symptoms after the age of 8 years (Reference AicardiAicardi, 1998), by which time all, or almost all, will exhibit a Kayser–Fleischer ring, a brown granular deposition of copper at the junction of the cornea and sclera. Psychiatric manifestations are often prominent and may constitute the sole or most prominent feature for months or years. Personality change, with irritability and a low threshold to anger, deteriorating academic or work performance and depression are the most common behavioural features, but the full spectrum of psychiatric disorders has been reported in some cases (Reference Akil, Brewer, Weiner and LangAkil & Brewer, 1995). The most common neurological manifestation in childhood is dystonia. Faciolingual–pharyngeal involvement, producing mask-like facies, dysarthria and dysphagia with impairment of speech, is usually the dominant feature, although chorea and myoclonic movements may also be observed.
Myoclonus
Myoclonus consists of brief, shock-like muscle jerks similar to those provoked by stimulating the nerve to a muscle with an electric shock. It differs from chorea in that there are distinct pauses between each movement and jerks occur repetitively in the same muscle (Reference MarsdenMarsden, 1998). In children, myoclonus may be a manifestation of primary generalised epilepsy or a particular epilepsy syndrome, or it might indicate a progressive degenerative disorder. Table 5 lists some of these disorders, two of which I discuss below.
Sub-acute sclerosing panencephalitis (SSPE) is caused by the measles virus and preferentially affects those who suffered wild measles infection in the first 2 years of life. It has declined in frequency since the introduction of the measles, mumps and rubella (MMR) vaccine. The median interval to SSPE is 8 years, with an insidious onset of personality change and subtle intellectual deterioration in 66% of cases (Reference AicardiAicardi, 1998). Within months, myoclonus occurs which is absent during sleep and which may disappear and reappear in wakefulness with no apparent explanation. Generalised seizures, vacant spells and partial seizures may all be seen. Over time, extrapyramidal dysfunction and spasticity evolve, along with dementia. The electroencephalogram is characteristic, with paroxysmal complexes of high-voltage slow waves which may be visible before any clinical manifestation.
Although rare at present, the number of cases of new variant Creutzfeldt–Jakob disease is increasing. Most cases present with early psychiatric symptoms of various types followed, after a median of 6 months, by ataxia (Reference Will, Zeidler and StewartWill et al, 2000) and, subsequently, by involuntary movements and cognitive impairment. In the cases reported from the National Surveillance Unit in Edinburgh (Reference Will, Zeidler and StewartWill et al, 2000), the median duration of the illness was 14 months. Since there is significant variation in the presenting psychiatric disturbance, a high index of suspicion is needed to identify these cases early.
Conclusions
There are many elements of paediatric neurological observation and examination which are of relevance to the child and adolescent psychiatrist (see Reference Dooley and GoldbloomDooley, 1997, for further advice on the technique of examination of children). Individuals with psychiatric morbidity, in whom neurological signs appear or progress, require closer examination. It is also important to examine children and young people presenting with behavioural change and, in particular, those in whom there is any suggestion of failure to acquire new abilities or in whom loss of abilities is reported. There is a child and adolescent psychiatry training module in the training programme of paediatric neurologists which I personally found very useful and enjoyable. Perhaps it would be beneficial to have a paediatric neurology module in the training programme for child psychiatrists.
Multiple choice questions
-
1 A diagnosis of neurofibromatosis type 1:
-
a is supported by the presence of 6 or more café-au-lait spots, >5 mm in children
-
b is supported by the presence of copious freckles
-
c cannot be sustained without a family history
-
d is supported by the presence of an optic glioma
-
e can be associated with specific learning difficulties affecting non-verbal skills more than verbal skills.
-
-
2 The diagnosis of tuberous sclerosis:
-
a is associated with the presence of subependymal glial nodules
-
b is rarely seen sporadically
-
c is infrequently associated with epilepsy
-
d is frequently associated with the presence of autistic features
-
e is characteristically associated with adenoma sebaceum.
-
-
3 The following diagnoses are associated with microcephaly:
-
a Sotos syndrome
-
b fragile-X syndrome
-
c Rett syndrome
-
d neurofibromatosis type 1
-
e Angelman syndrome.
-
-
4 The following ophthalmic features are associated with neurodegenerative disorders:
-
a static visual field defects
-
b optic atrophy
-
c Lisch nodules
-
d a cherry-red spot
-
e supranuclear ophthalmoplegia.
-
-
5 The following statements are true:
-
a tic disorder of childhood may be genetically inherited
-
b Huntington's chorea presents only in adulthood and after the diagnosis is known in the affected parent
-
c the search for the diagnosis of Wilson's disease is not cost-effective because the outcome cannot be altered
-
d new variant Creutzfeldt–Jakob disease may initially present with isolated neuropsychiatric disturbance
-
e an electroencephalogram should be performed in all children with progressive myoclonus, as it may be diagnostic.
-
MCQ answers

| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | T | a | T | a | F | a | F | a | T |
| b | F | b | F | b | F | b | T | b | F |
| c | F | c | F | c | T | c | F | c | F |
| d | T | d | T | d | F | d | T | d | T |
| e | T | e | T | e | T | e | T | e | T |






eLetters
No eLetters have been published for this article.