


Nearly 4% of all deaths worldwide are attributable to alcohol (WHO, 2011). In the United States, alcohol is the third leading preventable cause of death, after obesity and tobacco use (Mokdad et al., Reference Mokdad, Marks, Stroup and Gerberding2004). This public health burden is unevenly distributed across the life course with the highest levels of use, abuse, and dependence in early adulthood. It is well established that both alcohol use and alcohol use disorder rates increase markedly across adolescence and into the early twenties (Chen & Kandel, Reference Chen and Kandel1995; Harford et al., Reference Harford, Grant, Yi and Chen2005), after which disorder rates decrease considerably during young adulthood (Harford et al., Reference Harford, Grant, Yi and Chen2005). In the 2001 National Household Survey on Drug Abuse, 12-month prevalence rates for any alcohol disorders dropped 28% for males and 47% for females from ages 18–23 years to ages 24–29 years (Harford et al., Reference Harford, Grant, Yi and Chen2005). During early adulthood, alcohol is the leading cause of death worldwide (WHO, 2011). Clearly, adolescence and young adulthood are critical developmental stages for studying alcohol misuse and its genetic influences (Rutter et al., Reference Rutter, Silberg, O’Connor and Simonoff1999).
Across a number of measures (e.g., drinking frequency, binge drinking, alcohol abuse, or dependence), alcohol involvement is clearly familial and moderately heritable (h 2 ≈ 40–60%; Kendler et al., Reference Kendler, Schmitt, Aggen and Prescott2008; Miles et al., Reference Miles, Silberg, Pickens and Eaves2005; Pagan et al., Reference Pagan, Rose, Viken, Pulkkinen, Kaprio and Dick2006; Silberg et al., Reference Silberg, Rutter, D’Onofrio and Eaves2003). This finding has led to extensive genome-wide linkage and candidate gene studies to identify specific susceptibility markers. Indeed, a number of risk variants have been identified (e.g., acetaldehyde and alcohol dehydrogenase genes) (Quertemont, Reference Quertemont2004), but many others have failed to replicate these in independent samples. Genome-wide association studies (GWAS) offer comprehensive tests of common genetic variants and have proven successful in characterizing the genetic architecture of many complex diseases (The Wellcome Trust Case Control Consortium, 2007). To date, a small body of GWAS have studied problematic alcohol use and related endophenotypes and identified a number of putative risk loci (Bierut et al., Reference Bierut, Agrawal, Bucholz, Doheny, Laurie, Pugh and Rice2010; Edenberg et al., Reference Edenberg, Koller, Xuei, Wetherill, McClintick, Almasy and Foroud2010; Heath et al., Reference Heath, Whitfield, Martin, Pergadia, Goate, Lind and Montgomery2011; Schumann et al., Reference Schumann, Coin, Lourdusamy, Charoen, Berger, Stacey and Elliott2011; Treutlein et al., Reference Treutlein, Cichon, Ridinger, Wodarz, Soyka, Zill and Rietschel2009). Implicated variants include polymorphisms in PECR (Johnson et al., Reference Johnson, Drgon, Liu, Walther, Edenberg, Rice, Foroud and Uhl2006; Treutlein et al., Reference Treutlein, Cichon, Ridinger, Wodarz, Soyka, Zill and Rietschel2009), a gene cluster on chromosome 11 (Edenberg et al., Reference Edenberg, Koller, Xuei, Wetherill, McClintick, Almasy and Foroud2010), PKNOX2 (Bierut et al., Reference Bierut, Agrawal, Bucholz, Doheny, Laurie, Pugh and Rice2010), KIA0040 (Bierut et al., Reference Bierut, Agrawal, Bucholz, Doheny, Laurie, Pugh and Rice2010; Zuo et al., Reference Zuo, Gelernter, Zhang, Zhao, Lu, Kranzler and Luo2011), HTR1 (Bierut et al., Reference Bierut, Agrawal, Bucholz, Doheny, Laurie, Pugh and Rice2010; Zuo et al., Reference Zuo, Gelernter, Zhang, Zhao, Lu, Kranzler and Luo2011), and AUTS2 (Schumann et al., Reference Schumann, Coin, Lourdusamy, Charoen, Berger, Stacey and Elliott2011), among others. Particularly compelling evidence has been presented for rs6943555 in AUTS2, which was associated with alcohol consumption in a large meta-analysis (N ≈ 26K), and corroborated by AUTS2 expression data in human prefrontal cortex and murine brain, as well as reduced alcohol sensitivity with AUTS2 homolog downregulation in Drosophila. Such robust findings remain the exception to the rule; however, as most genetic variants implicated in GWAS studies have failed to replicate.
In response to the challenges of genomic studies of developmentally dynamic, difficult to measure substance abuse phenotypes, the NIDA-sponsored Genes, Environment and Development Initiative (GEDI) combines three longitudinal samples of psychiatric/behavioral repeated assessments with genome-wide data generated using a common platform (Illumina Human660W-Quad v1). The project offers several scientific advances. Foremost, harmonized data across three independent samples reduces the risk of study-specific artifacts, facilitating the search for cross-validated, replicated signals (Agrawal et al., Reference Agrawal, Freedman, Cheng, Lin, Shaffer, Sun and Beirut2012). Second, longitudinal data offer multiple statistical and substantive benefits (Adkins et al., Reference Adkins, Aberg, McClay, Hettema, Kornstein, Bukszar and van den Oord2010). Most fundamentally, statistical models of individual development are unidentifiable without multiple assessments per subject (Singer, Reference Singer2003). Thus, GEDI's structure allows genome-wide investigation of genetic risk for longitudinal patterns of alcohol abuse across its principle risk period –– adolescence and young adulthood. Next, and crucially, when measuring any outcome with non-trivial measurement error (e.g., alcohol consumption), increasing the number of assessments/subject monotonically increases estimate precision, and consequently statistical power, with dramatic gains when assessments/subject are numerous, as in GEDI (Willett et al., Reference Willett, Singer and Martin1998). Modeling all GEDI alcohol consumption assessments (>5 per subject) increases estimate precision >3-fold compared to using only first and last assessments (Willett et al., Reference Willett, Singer and Martin1998). Finally, we employ the simple, often overlooked, strategy of improving statistical power by specifying a quantitative outcome –– alcohol consumption. The power gains of quantitative versus dichotomous outcome specifications are well documented (Cohen, Reference Cohen1983), and it has been demonstrated that focusing solely on clinical diagnoses in psychiatric GWAS dramatically decreases power to detect effects (van der Sluis et al., Reference van der Sluis, Posthuma, Nivard, Verhage and Dolan2013).
Cumulatively, these techniques help maximize power in the current study, which analyzes three longitudinal samples with repeated assessment of alcohol consumption across adolescence and young adulthood (total N = 2,126, Obs = 12,166). Consumption-repeated measurements were analyzed using linear mixed models, estimating individual consumption trajectories (and a longitudinal mean consumption measure), which were then tested for association with Illumina 660W-Quad genome-wide data (866,099 SNPs after genotype imputation and QC). Association results were combined across samples using standard meta-analytic procedures (de Bakker et al., Reference de Bakker, Ferreira, Jia, Neale, Raychaudhuri and Voight2008). Top associations were examined for evidence of enriched association to known biological pathways.
Materials and Methods
Description of Individual Studies
Great smoky mountain study (GSMS)
GSMS is a longitudinal, representative study of children in 11 predominantly rural counties in southeast United States begun in 1993 (Costello et al., Reference Costello, Angold, Burns, Stangl, Tweed, Erkanli and Worthman1996). Three cohorts of children, aged 9, 11, and 13 years, were recruited from a pool of ~20,000 children, resulting in N = 1,420 participants (49% female). Annual assessments were completed with the child and primary caregiver until age 16 years and then with the participant again at ages 19, 21, and 24–26 years for 9,904 total assessments. These assessments used the Childhood and Adolescent Psychiatric Assessment (Angold & Costello, Reference Angold and Costello2000) and its upward extension. Informed consent forms were completed for all aspects of data collection and the study protocol was approved by the Duke Institutional Review Board. An average of 82% of all possible interviews was completed across all waves, ranging from 75% to 94% at individual waves. Blood spots collected at each observation were used for DNA extraction performed at Rutgers University Cell and DNA Repository. A total of 784 GSMS subjects, contributing 5,766 repeated alcohol consumption assessments, were analyzed in genome-wide association testing. Study characteristics are summarized in Table 1.
TABLE 1 Characteristics of the Studies

Christchurch health and development study (CHDS)
The CHDS is a longitudinal study of a birth cohort of 1,265 children born in the Christchurch region of New Zealand in 1977 (Fergusson & Horwood, Reference Fergusson and Horwood2001). This cohort involved 97% of children born from April 15 to August 5, 1977 and has been assessed on 22 occasions to age 30 years. Data were gathered during face-to-face interviews with subjects and parents, supplemented by data from official records. Signed consent was obtained for all aspects of data collection and the study has been subject to ethical review throughout its history. The present analysis is based on data collected during assessments of the cohort in adolescence (ages 14, 15, 16, and 18 years) and adulthood (ages 21, 25, 30 years). The number of subjects assessed at these ages ranged from 953 to 1,025, representing between 76% and 82% of the surviving cohort at each age. Whole blood was collected from most subjects, but for ~9% of participants saliva samples were used instead (Oragene TM, DNA Genotek Inc., Ontario, Canada). DNA extractions were performed at the University of Otago, Gene Structure, and Function Lab. A total of 739 CHDS subjects, contributing 4,959 repeated alcohol consumption assessments, were analyzed in genome-wide association testing.
Virginia twin study on adolescent behavioral development (VTSABD)
The VTSABD is an ongoing cohort-longitudinal study of twins born 1974–1983; 1,412 families were included in the first wave of data collection, with three subsequent waves of data collection occurring at approximately 1.5-year intervals, and a fifth wave when participants were in their mid-20s (Simonoff et al., Reference Simonoff, Pickles, Meyer, Silberg, Maes, Loeber and Eaves1997). A sample of 1,894 putative twin pairs was ascertained through the state school system and participating private schools in Virginia, and through families who contacted the VTSABD. A total of 1,412 families (2,775 children) participated (75%). Up to age 18 years, children and parents completed the Childhood and Adolescent Psychiatric Assessment. Parents completed a similar assessment on both twins. After the age of 18 years, the twins alone were interviewed using an age-updated interview designed for telephone administration. Whole blood was collected from VTSABD subjects and DNA extractions were completed at Rutgers University Cell and DNA Repository. A total of 603 unrelated VTSABD subjects, contributing 1,441 repeated alcohol consumption assessments, were analyzed in genome-wide association testing.
Genotyping and Quality Control
All samples were genotyped using Illumina Human660W-Quad v1 DNA Analysis BeadChips at the Mayo Clinic. Quality control procedures were applied to each study using R, PLINK (Purcell et al., Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira, Bender and Sham2007) and EIGENSOFT (Patterson et al., Reference Patterson, Price and Reich2006). In each study, single nucleotide polymorphisms (SNPs) with missing rate>0.01, minor allele frequency (MAF)<0.05, or extreme deviation (p <1E-6) from Hardy–Weinberg equilibrium were removed from further analysis. Subjects with missing rate >0.01 or unusual genome-wide homozygosity (|normalized homozygosity rate|>5) were excluded. Sex was investigated using no-call proportions of chr Y SNPs and heterozygosity proportions for chr X SNPs. Mislabeled sex information was corrected after double-checking the original data and subjects with unexplainable results were deleted. In addition, pairwise identical-by-descent (IBD) estimation was evaluated to identify unexpected duplicates and relative pairs. For VTSABD, one subject per twin pair, selected at random, was retained in the current analysis.
SNP dosages were imputed in each study using MaCH (Li et al., Reference Li, Willer, Ding, Scheet and Abecasis2010). The imputation reference was HapMap3 CEU (Utah residents with Northern and Western European ancestry) for subjects of European ancestry. Unobserved population admixture due to ancestry is a well-known confound in GWAS (Patterson et al., Reference Patterson, Price and Reich2006). To protect against false positives due to ancestry, we extracted five principal components from each sample to capture population stratification. To improve the efficiency of population stratification principal components analysis (PCA), a subset of independent SNPs was selected using PLINK, with 77155–79517 SNPs analyzed in each study. PCA was applied to the selected SNPs using the ‘smartpca’ module of EIGENSOFT.
Data Analysis
Phenotype Modeling
Two longitudinal measures of over-time alcohol consumption were used as phenotypic outcomes. Both measures were derived from repeated assessments of average drinks per week. The first measure, as detailed below, was a developmental trajectory estimate of alcohol consumption (drinks per week) across adolescence and early adulthood (maximum age range: 8–30 years). Intuitively, this trajectory slope measures the rate at which alcohol consumption increases, starting with no consumption for virtually all subjects in childhood, then increasing at different rates (or remaining flat among non-drinkers) through adolescence and the transition to adulthood, before stabilizing at different levels in the late twenties. The second measure was the simple mean of all alcohol consumption (drinks per week) repeated assessments collected across adolescence and the transition to adulthood (maximum age range: 12–21 years) for each subject. We selected these two specifications as: (1) the trajectory outcome was derived from the best fitting longitudinal model of the several tested, and maximized power through including all repeated measures, while (2) the mean consumption outcome provided a simpler summary of individuals’ drinking behavior, and thus greater continuity to existing literature (Agrawal et al., Reference Agrawal, Freedman, Cheng, Lin, Shaffer, Sun and Beirut2012; Grant et al., Reference Grant, Agrawal, Bucholz, Madden, Pergadia, Nelson and Heath2009). The mean consumption measure also has the benefit of focusing solely on adolescence and the transition to adulthood –– a developmental period of non-normative drinking, which is associated with increased risk of concurrent comorbid psychiatric disorders and future substance abuse and dependence (Rutter et al., Reference Rutter, Silberg, O’Connor and Simonoff1999).
To generate the developmental trajectory estimates, we applied a method to estimate longitudinal development from all available repeated observations using linear mixed models (van den Oord et al., Reference van den Oord, Adkins, McClay, Lieberman and Sullivan2009). This method first determines the optimal functional form of the developmental trajectory, then generates individual trajectory estimates based on the best-fitting model, using best linear unbiased predictors (BLUPs; Pinheiro & Bates, Reference Pinheiro and Bates2000). As this approach takes advantage of all available repeated assessments, it results in more precise estimates than traditional approaches that estimate development using two assessments (i.e., change scores; Willett et al., Reference Willett, Singer and Martin1998). Further, as estimates are based on mixed model trajectory slopes, they are more robust to attrition than competing approaches (Laird & Ware, Reference Laird and Ware1982). This method of estimating outcomes from longitudinal phenotypic assessments is well established in psychiatric GWAS (Adkins et al., Reference Adkins, Aberg, McClay, Bukszar, Zhao, Jia and van den Oord2011; Clark et al., Reference Clark, Adkins, Aberg, Hettema, McClay, Souza and van den Oord2012; McClay et al., Reference McClay, Adkins, Aberg, Bukszar, Khachane, Keefe and van den Oord2011a).
In modeling the developmental trajectories, we tested several specifications of age-based longitudinal change, including linear and quadratic functions, as well as a series of piecewise plateau models specifying linear change until a given age and flat thereafter (Bollen & Curran, Reference Bollen and Curran2005). Model comparisons using likelihood ratio tests for nested models, and BIC/AIC for non-nested quadratic and piecewise models (Burnham & Anderson, Reference Burnham and Anderson2004), indicated that a piecewise model with alcohol consumption stabilizing at approximately age 28 years best fit the data. Thus, the preferred model used to generate our trajectory slope outcome can be written as
 \begin{equation*}
Y_{ti} = \beta _{00} + \beta _{10} a _{ti} + u_{0i} + u_{1i} a_{ti} + \varepsilon _{ti} ,
\end{equation*}
\begin{equation*}
Y_{ti} = \beta _{00} + \beta _{10} a _{ti} + u_{0i} + u_{1i} a_{ti} + \varepsilon _{ti} ,
\end{equation*}
where i and t are subject and assessment occasions, respectively; Y is the alcohol consumption for subject i at assessment t; β 00 is the overall sample intercept; β 10 is the mean age slope coefficient for age specification a, which is coded 0 for the earliest age observed in the sample, 1 for second earliest age, and so forth, until reaching the plateau age (~28 years), after which its value remains constant for subsequent ages; u0i is the subject-specific deviation from that overall sample intercept; u1i is the subject-specific deviation from the mean age slope; ε ti is the residual for subject i at assessment t. Random effects u 0 and u1i were specified orthogonal. The term of interest, u1i , may more intuitively be described as the subject-specific alcohol consumption age trajectory. As subject-level parameters are not directly estimated by the mixed model, they were calculated as BLUPs in a post-estimation step (Pinheiro & Bates, Reference Pinheiro and Bates2000). See Supplementary Material 1 for further phenotypic modeling details.
Genome-Wide Association and Meta-Analyses
Genome-wide association testing was performed using PROBABEL (Aulchenko et al., Reference Aulchenko, Struchalin and van Duijn2010), GWAS software for use with probabilistically imputed genotype dosages. Tests were specified as linear regressions of additive SNP effects, controlling for the top five ancestral PCA dimensions as covariates. Meta-analysis procedures proposed by de Bakker et al. (Reference de Bakker, Ferreira, Jia, Neale, Raychaudhuri and Voight2008) were used to aggregate results across the three studies. Specifically, aggregate β coefficients and associated standard errors were computed as follows:
 \begin{equation*}
\begin{array}{l}
\beta = \displaystyle\frac{{\sum\nolimits_i {\left[ {\beta _i /(SE_i )^2 } \right]} }}{{\sum\nolimits_i {\left[ {1/(SE_i )^2 } \right]} }} \\[12pt]
SE = \sqrt {\displaystyle\frac{1}{{\sum\nolimits_i {\left[ {1/(SE_i )^2 } \right]} }}} ,
\end{array}
\end{equation*}
\begin{equation*}
\begin{array}{l}
\beta = \displaystyle\frac{{\sum\nolimits_i {\left[ {\beta _i /(SE_i )^2 } \right]} }}{{\sum\nolimits_i {\left[ {1/(SE_i )^2 } \right]} }} \\[12pt]
SE = \sqrt {\displaystyle\frac{1}{{\sum\nolimits_i {\left[ {1/(SE_i )^2 } \right]} }}} ,
\end{array}
\end{equation*}
where i indexes study number. Given the use of a common genotyping platform (Illumina Human660W-Quad v1 DNA Analysis BeadChips) and genotyping lab (Mayo Clinic), many common pitfalls in GWAS meta-analysis were avoided, including strand orientation mismatches, imputation reference sample heterogeneity, variable QC criteria, and so forth.
Multiple Testing
We used a false discovery rate (FDR; Benjamini & Hochberg, Reference Benjamini and Hochberg1995) approach to declare statistical significance. In comparison to controlling a family-wise error rate (e.g., Bonferroni correction), FDR: (a) provides a better balance between finding true effects versus controlling false discoveries, (b) results in comparable standards for declaring significance across studies because it does not directly depend on the number of tests, and (c) is relatively robust against having correlated tests (Brown & Russell, Reference Brown and Russell1997). FDR is commonly used in high-dimensional applications, including GWAS (Beecham et al., Reference Beecham, Martin, Li, Slifer, Gilbert, Haines and Pericak-Vance2009; Dubois et al., Reference Dubois, Trynka, Franke, Hunt, Romanos, Curtotti and van Heel2010; McClay et al., Reference McClay, Adkins, Aberg, Stroup, Perkins, Vladimirov and van den Oord2011b). We set an FDR threshold of 0.1 for declaring genome-wide significance. This specifies that, on average, 10% of the SNPs declared significant are expected to be false discoveries. Additionally, we discuss suggestive associations at an FDR threshold of 0.2 to reduce the probability of Type II statistical errors, while explicitly noting reduced confidence in these associations. Operationally, FDR was controlled using q-values, which are FDRs calculated using the p-value of the markers as thresholds for declaring significance (Storey & Tibshirani, Reference Storey and Tibshirani2003).
Pathway Analyses
Beyond our primary meta-analysis of single marker GWAS findings, we also conducted secondary analyses to determine whether any known biological pathways harbored an excess of SNPs with small p-values. Specifically, to interrogate the data for evidence of enriched association to known biological pathways, we conducted pathway analysis using ConsensusPathDB (http://cpdb.molgen.mpg.de/), a human-centric meta-database of functional biological data, compiled from 30 separate public sources of biological interactions (Kamburov et al., Reference Kamburov, Pentchev, Galicka, Wierling, Lehrach and Herwig2011). All alcohol consumption-associated SNPs with meta-analysis p < .01 were matched to the closest gene ±25 kb using RefSeq (GRCh37) coordinate information. All implicated genes were assembled into a final non-redundant gene list, comprising 2,590 (mean consumption) and 2,506 (developmental trajectory) genes. For each of the 4,601 reference pathways present in ConsensusPathDB, a hypergeometric test was performed to calculate the significance of the overlap between the genes from our putative susceptibility list and those present in each reference pathway. FDR was implemented to adjust significance for the large number of tests performed.
Results
Single Marker GWAS
Table 2 provides details on those SNPs that were genome-wide significant/suggestive (q < 0.1/q < 0.2). Figure 1A shows Q–Q plots for meta-analyses. The plots show that the p-values from the GWAS are generally on a straight line, indicating that the p-value distribution generally conforms to the expected null distribution assuming no effects of the markers. However, in both plots, there is also evidence that markers in the right upper corner have p-values smaller than would be expected under the null hypothesis, suggesting true association between these markers and the outcome. The plots also display λ values (i.e., ratio of median observed p-value to the median expected p-value under the null hypothesis) approximately equal to 1 (λ = 0.998 and 1.000), indicating no systematic test statistic inflation and adequate control of population stratification (see Supplementary Material 2 for study-specific Q–Q plots). Manhattan plots are provided in Figure 1B, and provide a low-resolution visualization of spatial clustering among association signals. The full set of GWAS p-values considered in this study is available for download at: http://www.pharmacy.vcu.edu/biomarker/resources/supplementary/.
TABLE 2 Strongest SNP-Longitudinal Alcohol Consumption Associations (q < 0.2)


FIGURE 1 (A) QQ plots and (B) Manhattan plots for GWAS results of two longitudinal alcohol consumption measures.
The top significant finding involves three genome-wide significant SNPs (rs7031417: q = 0.01, p = 1.4E-08; rs17053864: q = 0.05, p = 1.5E-07; rs7019589: q = 0.06, p = 2.3E-07) at pseudogene LOC100129340 (mitofusin-1-like) associated with mean adolescent alcohol consumption. These SNPs show moderately low MAF (~0.09) with minimal MAF differences across the three samples (Table 2). Study specific results show that this meta-analysis association was not driven by a single sample, with GSMS and CHDS exhibiting comparably low p-values (Table 2). Examination of the regional plot for the locus shows a robustly associated haplotype spanning ~100 kb, with >10 spatially clustered SNPs showing nominal association (Supplementary Material 3A). Notably, one of the genome-wide significant hits is located in the transcribed exon 4.
The second genome-wide significant association, also to mean consumption, is a locus in GABA transporter SLC6A1 (solute carrier family 6 (neurotransmitter transporter, GABA), member 1). In addition to the genome-wide hit in this gene (rs11710497: q = 0.05, p = 1.2E-07), a second SNP met our suggestive criteria (rs6778281: q = 0.15, p = 7.1E-07). Again, MAFs were moderately low (~0.10) and highly comparable across individual samples. Also, significance was not driven by a single study, with comparably low p-values for GSMS and VTSABD (Table 2). Inspection of the regional plot indicates a fairly small LD block overlaying exonic regions of SLC6A1. Further, this small linkage disequilibrium (LD) block is highly enriched for strong associations including several nominally associated coding SNPs (p < .001; Supplementary Material 3B).
Top findings for the developmental consumption trajectory included a cluster of correlated SNPs about 150 kb upstream of ADRA2A (adrenoceptor alpha 2A), with rs12257178 exhibiting the strongest association (q = 0.14, p = 4.8E-07). MAFs were high (~0.4) and homogenous across studies. Association strength was primarily driven by GSMS and CHDS samples (Table 2). While it is questionable whether this putative susceptibility locus is in LD with ADRA2A, the regional plot does show a correlated SNP located ~80 kb upstream of the gene (Supplementary Material 3C). Also, further inspection of the regional plot indicates an unusually wide area of moderately enriched association surrounding the suggestive locus, spanning ~500 kb and encompassing ADRA2A.Thus, it is possible that this association may tag cis regulatory elements or genic variants for ADRA2A.
Other top hits included a pair of SNPs in high LD (R 2>0.9) within ZNF578 (rs1984450 and rs7253326: q = 0.14; p ≈ 2.7E-07) associated with the trajectory outcome, and an interesting spatial pattern of SNPS associated with mean consumption surrounding an intronic SNP in MIPOL1 (rs4898641: q = 0.19, p = 1.1E-06). The ZNF578 finding exhibits almost exactly the same level of association across studies (p ≈ 3.0E-03) and moderately low MAF (~0.10). Correlated signals overlapped ZNF578 and the adjacent ZNF808 and ZNF701 genes, and tag two nominally significant coding SNPs in ZNF578 (Supplementary Material 3D). Next, rs4898641 in MIPOL1 appears to tag a ~200 kb haplotype, enriched for association, which closely corresponds to the gene boundaries of MIPOL1 (Supplementary Material 3E). Our final suggestive finding involved a genic locus in IGSF9B centered on rs694424 (q = 0.14, p = 5.4E-07; Supplementary Material 3F). This finding is of lower confidence, however, as its association strength is essentially driven by a single sample (VTSABD).
Pathway Analyses
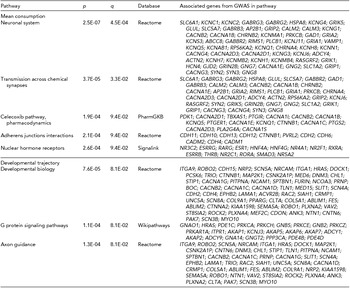
Results from ConsensusPathDB indicated significant involvement of several intuitive biological pathways (Table 3). As ConsensusPathDB is a meta-database bioinformatic tool, it mines multiple individual databases –– in the current analysis significant (q < 0.1) pathways were found in Reactome, PharmGKB, Signlink, and Wikipathways databases (Kamburov et al., Reference Kamburov, Pentchev, Galicka, Wierling, Lehrach and Herwig2011). The top pathways, ‘neuronal system’ (p = 2.5E-07, q = 4.5E-04) and the related ‘transmission across chemical synapses’ (p = 3.7E-05, q = 3.3E-02), indicated an association between genetic variation in neurotransmission and individual differences in mean adolescent alcohol consumption, with disproportionate representation of GABAergic and glutamatergic genes in both of these pathways. Other significant pathways for mean adolescent consumption including ‘NHR’ (p = 2.6E-04, q = 9.4E-02), ‘celecoxib pharmacodynamics’, and ‘adherens junctions interactions’ (p = 2.1E-04, q = 9.4E-02; Table 3). The three significant pathways for the developmental consumption trajectory indicated more general developmental themes, with specific findings including ‘developmental biology’ (p = 7.6E-05, q = 8.1E-02), ‘G protein signaling pathways’ (p = 1.1E-04, q = 8.1E-02) and ‘axon guidance’ (p = 1.3E-04, q = 8.1E-02).
TABLE 3 ConsensusPathDB Pathway Results for All SNPs Within Genes Nominally Associated (p < .01) With Longitudinal Alcohol Consumption
Discussion
Developmental perspectives have long stressed the importance of accounting for temporality and life course variation in models of substance abuse. A primary insight of such perspectives is that the importance of various risk factors fluctuates across developmental trajectories (Adkins et al., Reference Adkins, Daw, McClay and Van den Oord2012; Willett et al., Reference Willett, Singer and Martin1998). The current study endeavors to wed this perspective to genomic approaches to alcohol consumption. While psychiatric genetics has made advances toward elucidating the link between genetic variation and alcohol involvement, this research has largely been atemporal. The weakness of such static perspectives on the genetic determinants of alcohol involvement is highlighted not only by developmental perspectives, but also by research within genetics showing that epigenetic mechanisms (i.e., methylation, histone modification, and chromatin remodeling) regulate gene expression in response to developmental and environmental cues (Whitelaw & Whitelaw, Reference Whitelaw and Whitelaw2006). Using the GEDI GWAS samples, this longitudinal GWAS meta-analysis has addressed the issue of developmental variation in genetic influences across adolescence and young adulthood through genome-wide testing of common variant effects on longitudinal measures of alcohol consumption, employing FDR methods to control the risk of false discoveries.
Our top genome-wide finding involved three SNPs (rs7031417, rs17053864, rs7019589) at pseudogene LOC100129340 (mitofusin-1-like). Very little is known regarding this pseudogene, which was only added in the most recent, hg19, gene track (thus, its absence in the regional plot). However, variants within LOC100129340 were found to be associated with adult human height in a large GWAS meta-analysis (Gudbjartsson et al., Reference Gudbjartsson, Walters, Thorleifsson, Stefansson, Halldorsson, Zusmanovich and Stefansson2008). Also, one of the genome-wide significant hits fell within exon 4 of the pseudogene. Given that pseudogenes, by definition, lack protein-coding function, the prior probability of this locus being a true discovery is somewhat diminished. However, considering (a) the association signal is in a transcribed sequence, (b) pseudogene transcripts have demonstrated trans-regulation of homologous coding genes (Hirotsune et al., Reference Hirotsune, Yoshida, Chen, Garrett, Sugiyama, Takahashi and Yoshiki2003), and (c) endogenous siRNA are sometimes derived from pseudogene transcription (Tam et al., Reference Tam, Aravin, Stein, Girard, Murchison, Cheloufi and Hannon2008), it would be rash to dismiss this result as a false discovery.
Our second genome-wide significant locus was tagged by rs11710497, located within GABA transporter 1—SLC6A1 (solute carrier family 6 (neurotransmitter transporter, GABA), member 1). Given that alcohol functions primarily through binding to GABA receptors, and that this gene encodes protein GAT-1, which removes GABA from the synaptic cleft, there is a notably high prior probability of this being a true discovery. Furthermore, while the two genome-wide significant/suggestive hits were in intronic regions, they tag strongly associated exonic variants in SLC6A1. Although the network biology of the gene suggests obvious links to alcohol consumption, there has been surprisingly little research studying this relationship. Conversely, studies have implicated the gene in anxiety disorders and ADHD (e.g., Lasky-Su et al., Reference Lasky-Su, Neale, Franke, Anney, Zhou, Maller and Faraone2008; Thoeringer et al., Reference Thoeringer, Ripke, Unschuld, Lucae, Ising, Bettecken and Erhardt2009; Reference Thoeringer, Erhardt, Sillaber, Mueller, Ohl, Holsboer and Keck2010). This finding should focus future research on the study of SLC6A1 variants in alcohol abuse and other psychiatric phenotypes related to cortical excitability/inhibition.
The strong association of a large region near ADRA2A also provides a promising potential biological mechanism. Alpha-2-adrenergic receptors are members of the G protein-coupled receptor superfamily. These receptors have a critical role in regulating neurotransmitter release from sympathetic nerves and from adrenergic neurons in the CNS. Alpha2A and alpha2C subtypes are required for normal presynaptic control of transmitter release from sympathetic nerves in central noradrenergic neurons, and the alpha2A subtype inhibits transmitter release at high neural stimulation frequencies. This gene encodes alpha2A subtype and it contains no introns in either its coding or untranslated sequences. There is an extensive literature on the gene and many findings suggesting association to ADHD (e.g., Kim et al., Reference Kim, Kim, Kang, Cho, Shin, Yoo, Hong and Lee2010; Roman et al., Reference Roman, Schmitz, Polanczyk, Eizirik, Rohde and Hutz2003; Reference Roman, Polanczyk, Zeni, Genro, Rohde and Hutz2005; Waldman et al., Reference Waldman, Nigg, Gizer, Park, Rappley and Friderici2006), as well as metabolic traits (e.g., glycemic phenotypes, blood pressure; for meta-analysis, see Talmud et al., Reference Talmud, Cooper, Gaunt, Holmes, Shah, Palmen and Humphries2011), and substance abuse (Clarke et al., Reference Clarke, Dempster, Docherty, Desrivieres, Lourdsamy, Wodarz and Schumann2012; Merenakk et al., Reference Merenakk, Maestu, Nordquist, Parik, Oreland, Loit and Harro2011; Prestes et al., Reference Prestes, Marques, Hutz, Roman and Bau2007). Given the neurobiological implications, this gene should be considered a strong candidate in future alcohol abuse studies. The final three genome-wide suggestive (q < 0.2) loci were for intronic SNPs in ZNF578, MIPOL1, and IGSF9B and have no reported evidence linking them to alcohol use/abuse phenotypes. Thus, future research will be required to adjudicate whether they are novel susceptibility loci or false discoveries.
Pathway analyses indicated several intuitive biological mechanisms. Top results for mean adolescent consumption (‘neuronal system’ and ‘transmission across chemical synapses’) suggested extensive involvement of GABAergic (e.g., SLC6A1, GABRG3, GABRG2, GABBR2) and glutamatergic (e.g., GRIK5, GLUL, GRIK1, GRIN2B) neurotransmission. Given that GABA and glutamate neurotransmitter systems are primary pharmacodynamic targets of alcohol (for review, see Vengeliene et al., Reference Vengeliene, Bilbao, Molander and Spanagel2008), these results are consistent with extant knowledge of the drug's biological mechanisms. ‘Celecoxib pharmacodynamics’ and ‘NHRs’ also offer intriguing biological mechanisms for genetic variation in alcohol metabolism. Celecoxib acts by inhibiting prostaglandin synthesis (Penning et al., Reference Penning, Talley, Bertenshaw, Carter, Collins, Docter and Isakson1997). Prostaglandins are lipid autocrine/paracrine mediators with a variety of physiological effects, including inflammation modulation (Hata & Breyer, Reference Hata and Breyer2004), which have been shown to moderate alcohol ‘hangover’ intensity (Wiese et al., Reference Wiese, Shlipak and Browner2000) and reduce alcohol-induced liver toxicity (Nanji et al., Reference Nanji, Khettry, Sadrzadeh and Yamanaka1993). ‘NHR, are intracellular proteins responsible for sensing steroid and thyroid hormones and some other signaling molecules. Multiple NHR genes implicated in the current analysis are specifically expressed in liver (e.g., HNF4A, HNF4G) and several others function as thyroid hormone receptors, which have been linked to severity of alcohol craving, consumption, and withdrawal (Alfos et al., Reference Alfos, Higueret, Pallet, Higueret, Garcin and Jaffard1996; Ozsoy et al., Reference Ozsoy, Esel, Izgi and Sofuoglu2006).
The three biological pathways significantly associated with the developmental consumption trajectory were broader and more explicitly developmentally oriented, particularly ‘developmental biology’ and ‘axon guidance’, which shared several implicated genes related to nervous system development (e.g., ROBO1, ROBO2, ANK3). The final significant pathway associated with the developmental trajectory, G-protein signaling, functions as an intermediary messenger system for many hormones, neurotransmitters, and other signaling molecules. While G-protein signaling is involved in many biological processes, it is known that alcohol intoxication is directly connected to the actions of four G-protein gated inwardly-rectifying potassium (GIRK) channel subunits, one of which was among the genes implicated in this pathway analysis (i.e., KCNJ3; Aryal et al., Reference Aryal, Dvir, Choe and Slesinger2009; Lewohl et al., Reference Lewohl, Wilson, Mayfield, Brozowski, Morrisett and Harris1999). Despite the general plausibility of these pathway results, it is worth noting that given the incomplete, low-resolution nature of current biological databases, findings should be regarded as tentative pending future tests of replication as database content and accuracy continue to improve (Khatri et al., Reference Khatri, Sirota and Butte2012).
Clearly, it is premature to suggest direct clinical applications of these findings for therapeutic target or biomarker discovery. On the contrary, actualizing the promise of translational genomics and converting academic findings into clinical applications will require a cumulative process of aggregating and jointly considering large bodies of evidence using meta-analytic and data integration techniques. Thus, it is important to rapidly disseminate genome-wide association results from longitudinal behavioral studies with genomic data, such as the GEDI samples. To facilitate this process, we provide all p-values (http://www.pharmacy.vcu.edu/biomarker/resources/supplementary/) as a resource for investigators with the requisite samples to carry out replication or further meta-analysis.
As with any genetic associations, our findings will require additional replication and functional validation. However, the present study shows the potential of GWAS meta-analyses of longitudinal data to discover genes and pathways that mediate developmental trends in alcohol consumption. A better understanding of these biological mechanisms and the roles of specific polymorphisms may facilitate the development of improved biomarker-based approaches to personalize substance abuse treatment. It is hoped that this research will eventually contribute to reducing the global health burden of substance abuse by facilitating identification of novel mechanisms, biomarkers, and therapeutic targets.
Acknowledgments
We would like to thank to the study participants from the Great Smoky Mountain Study, Virginia Twin Study on Adolescent Behavioral Development, and Christchurch Health and Development Study. Also, thanks to Karolina Aberg and colleagues at the VCU Center for Biomarker Research and Precision Medicine for helpful discussion and sharing analytical resources. Dr Conway’s role on this paper is as a Science Officer on U01DA014413, with no involvement in other cited grants. The views and opinions expressed in this report are those of the authors and should not be construed to represent the views of NIDA or any of the sponsoring organizations, agencies, or the US government. This research was supported by the National Institute on Drug Abuse (U01DA024413, R01DA11301), National Institute of Mental Health (R01MH063970, R01MH063671, R01MH048085, K01MH093731, and K23MH080230), NARSAD, and the William T. Grant Foundation. The CHDS was supported by the Health Research Council of New Zealand, National Child Health Research Foundation, Canterbury Medical Research Foundation, New Zealand Lottery Grants Board, University of Otago, Carney Centre for Pharmacogenomics, James Hume Bequest Fund, and NIH grant MH077874.
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/thg.2015.36





