

Introduction
MDM2 (Mouse Double Minute 2) inhibitors, which activate p53 by inhibiting MDM2-mediated p53 degradation, are under development for the treatment of TP53 wild-type cancer [Reference Tisato1]. The MDM2 inhibitor idasanutlin is currently investigated in clinical phase II and III trials for acute myeloid leukaemia (AML; NCT02670044, NCT02545283).
Resistance formation of TP53 wild-type cancer cells to MDM2 inhibitors commonly results in the formation of TP53 mutations as resistance mechanism [Reference Aziz2-Reference Jung8]. TP53 mutations may be the consequence of the selection of pre-existing TP53-mutant cell subpopulations or the induction of de novo TP53 mutations [Reference Michaelis3,Reference Drummond5-Reference Michaelis7]. De novo TP53 mutations may be the consequence of the selection of cells in which TP53 mutations have occurred by chance and which would have disappeared in the absence of the selection pressure induced by an MDM2 inhibitor. However, MDM2 inhibitors may also actively promote the formation of TP53 mutations by inducing DNA damage [Reference Vassilev9-Reference Wang12].
Objective
We used the AML cell line PL21 to investigate whether MDM2 inhibitor-induced DNA damage may promote the formation of TP53 mutations in the absence of a selection pressure. PL21 AML cells are TP53 wild-type (Table 1) but intrinsically resistant to nutlin-3 (an MDM2 inhibitor closely related to idasanutlin [Reference Vassilev9]), as indicated by a nutlin-3 IC50 of 20.49 μM (Figure 1, Table 1). Nutlin-3-sensitive cells display nutlin-3 IC50 values in the very low micromolar range, while nutlin-3 concentrations above 20 μM are associated with non-specific, p53-independent effects [Reference Michaelis3,Reference Michaelis7]. Twelve PL21 sublines were cultivated for 52 passages in the presence of nutlin-3 10 μM. The emergence of TP53 mutations in response to nutlin-3 treatment would indicate mutagenic effects that promote the formation of TP53 mutations also in the absence of a selective pressure on p53.
Table 1. Drug concentrations that reduce the viability of PL21 and its sublines cultivated for 52 weeks in the presence of nutlin-3 (20 μM) by 50% (IC50) as indicated by MTT assay after 120 h of incubation.
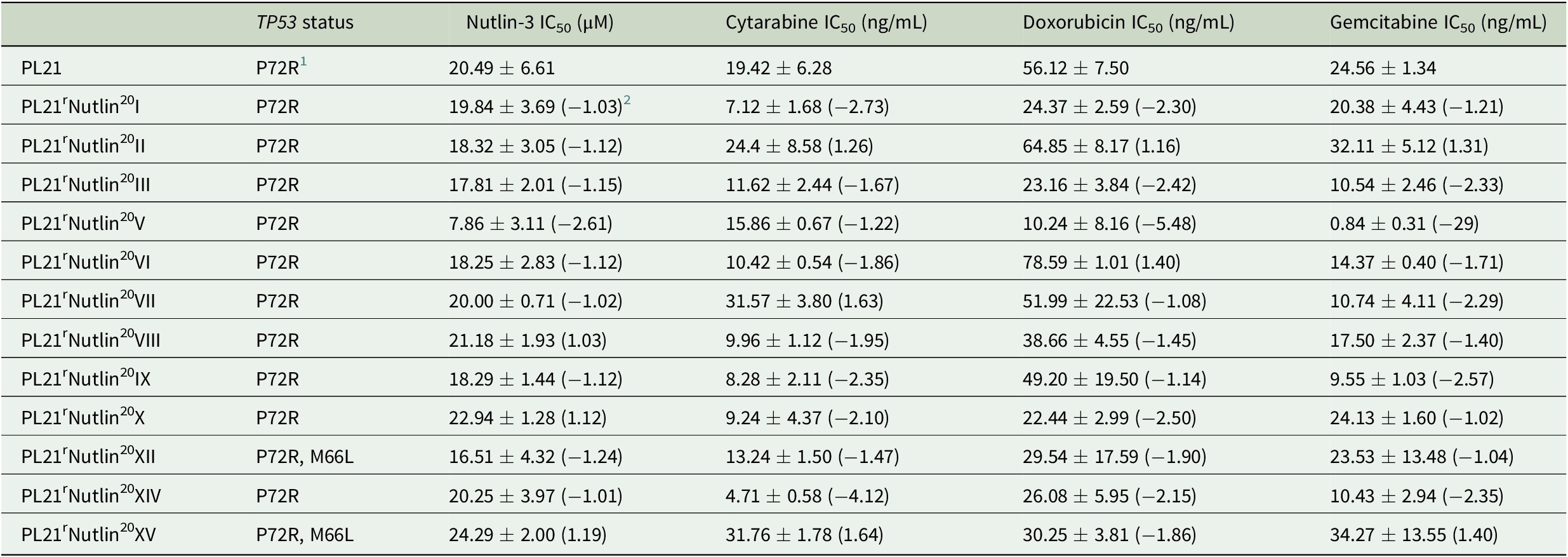
1 Polymorphism that does not affect p53 function.
2 Fold change relative to PL21.
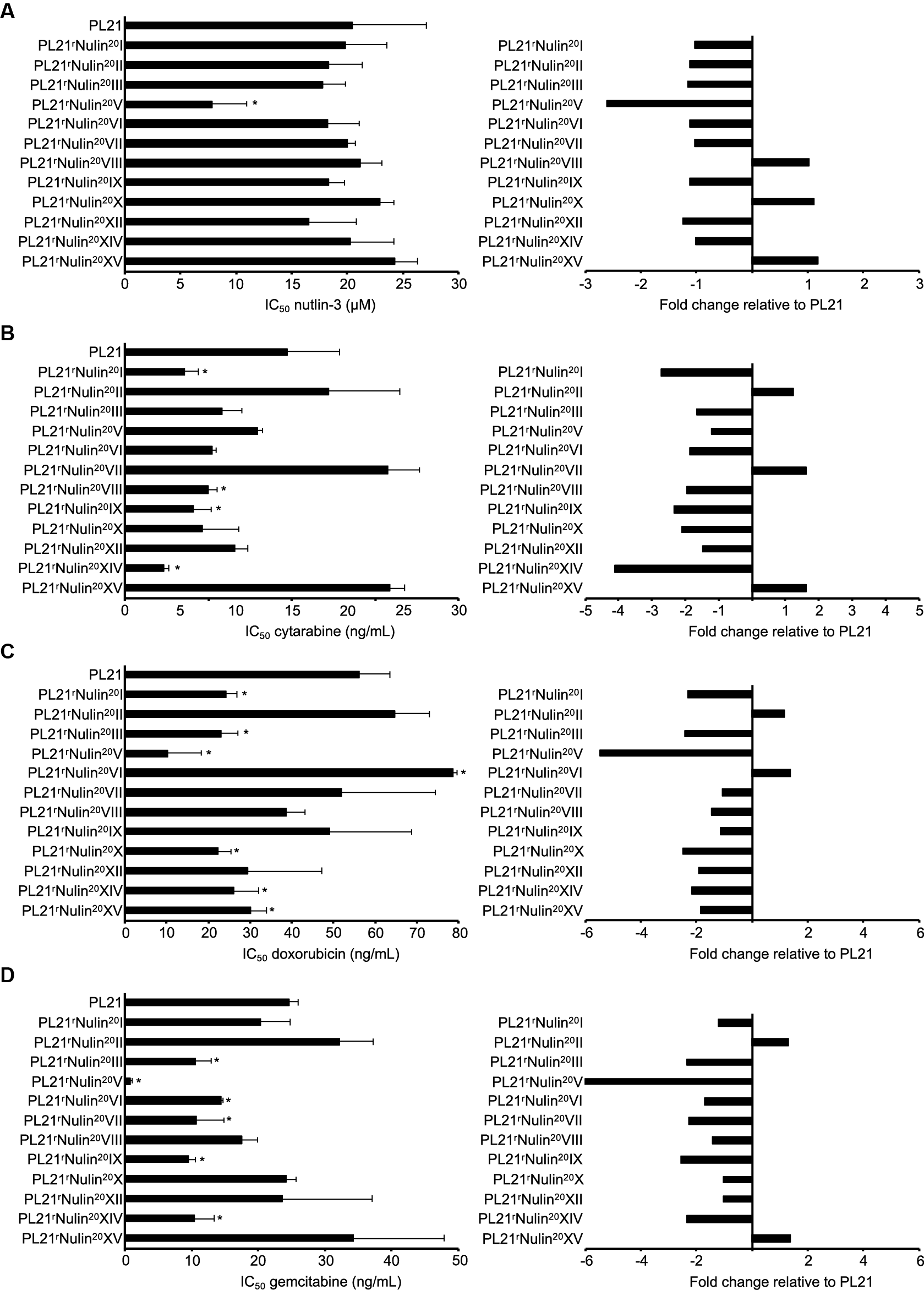
Figure 1. Drug sensitivity profiles of the AML cell line PL21 and its sublines cultivated in the presence of nutlin-3 (10 μM) for 52 weeks. Concentrations that inhibit cell viability by 50% (IC50, mean ± SD from three independent experiments) as determined by MTT assay after 120 h incubation and IC50 fold changes relative to PL21 were determined for nutlin-3 (A), cytarabine (B), doxorubicin (C), and gemcitabine (D). * P < 0.05 relative to PL21.
Methods
PL21 cells (DSMZ, Braunschweig, Germany) were cultivated in the absence or presence of drug in Iscove’s modified Dulbecco’s medium supplemented with 10% foetal calf serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin at 37 °C. Cells were routinely tested for mycoplasma contamination and authenticated by short tandem repeat profiling.
The TP53 status was determined by next generation sequencing, and cell viability was measured using eight drug concentrations by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [Reference Michaelis3,Reference Michaelis7]. Based on the MTT data, concentrations that inhibit cell viability by 50% (IC50) were determined using CalcuSyn (Biosoft, Cambridge, UK). Nutlin-3 was purchased from Selleck Chemicals via BIOZOL GmbH (Eching, Germany). Cytarabine was obtained from Tocris via Bio-Techne GmbH (Wiesbaden, Germany). Doxorubicin and gemcitabine were purchased from Teva GmbH (Ulm, Germany).
Results
All sublines had retained wild-type TP53 except for PL21rNutlin20XII and PL21rNutlin20XV, which displayed an M66L variant (Table 1). This variant was present in 386 (3.2%) out of 11,945 reads from the parental cell line and, hence not a de novo mutation induced by nutlin-3 treatment. If it had been of functional relevance, it would have been consistently selected by nutlin-3 treatment, as previously shown in other cell lines [5-7]. Thus, this observation does not suggest that nutlin-3 may directly induce TP53 mutations.
The 12 nutlin-3-treated PL21 sublines displayed an up to 3.1-fold variation in their nutlin-3 sensitivity (Figure 1, Table 1) and in their sensitivity to cytarabine (up to 6.7-fold), doxorubicin (up to 7.7-fold), and gemcitabine (up to 40.8-fold). Twelve PL21 sublines that had been cultivated for 52 weeks as control in parallel in the absence of nutlin-3 did not display any changes in their drug sensitivity profiles (Table 2).
Table 2. Drug concentrations that reduce the viability of PL21 sublines cultivated separately for 52 weeks by 50% (IC50) as indicated by MTT assay after 120 h of incubation. Values are represented as means ± S.D. of at least three independent experiments.
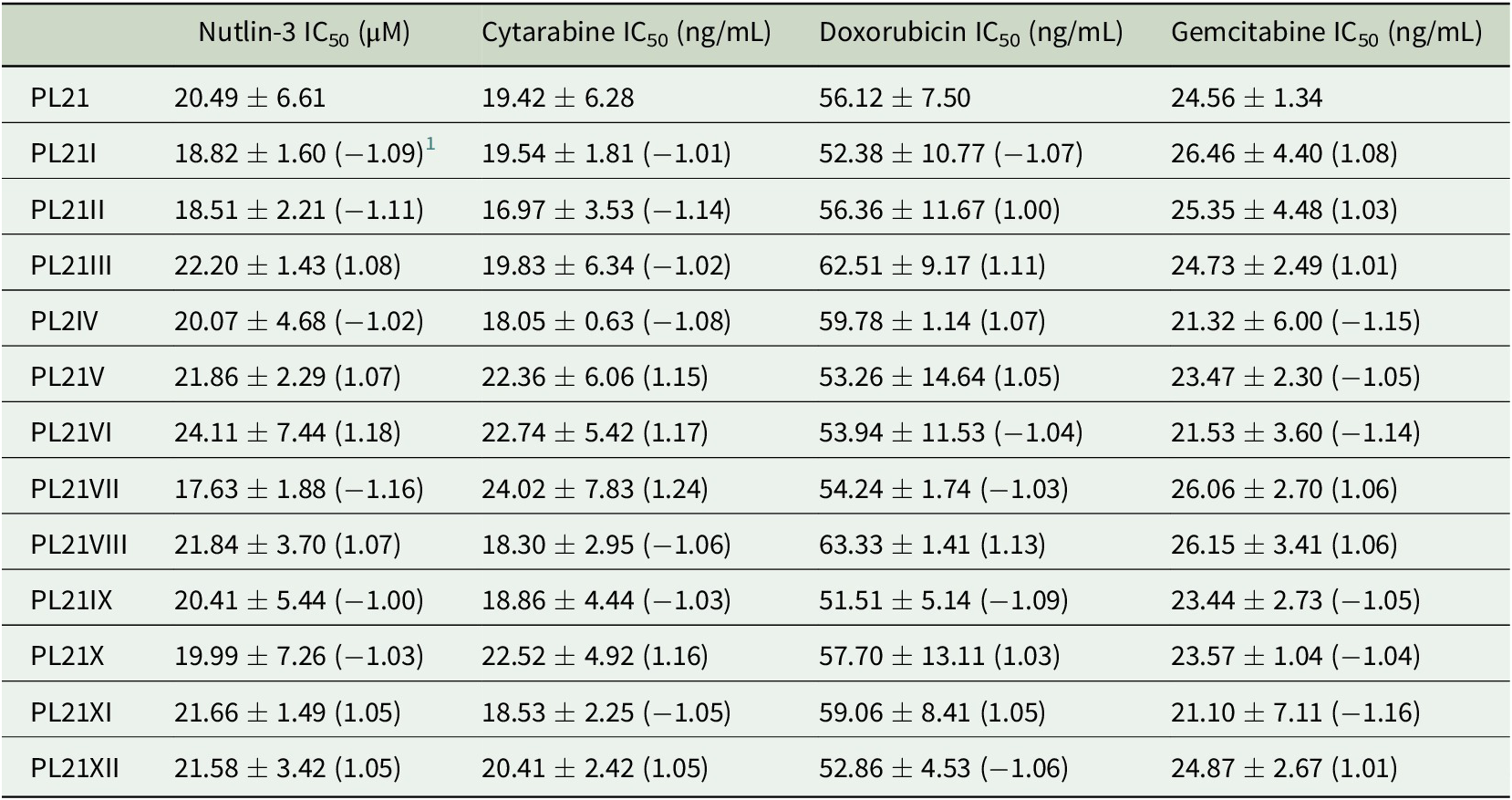
1 Fold change relative to PL21.
Discussion
Since treatment of PL21 cells with ineffective nutlin-3 concentrations did not result in loss-of-function TP53 mutations, TP53 mutations in MDM2 inhibitor-adapted cells may be rather the consequence of selection processes than of drug-induced mutations. In agreement, a fraction of MDM2 inhibitor-adapted cell lines retains wild-type TP53 [Reference Michaelis3,Reference Michaelis7]. Unexpectedly, prolonged nutlin-3 treatment resulted in increased sensitivity of a fraction of sublines to cytarabine, doxorubicin, or gemcitabine. In this context, MDM2 inhibition has been shown to increase the cellular reactive oxygen species (ROS) levels [Reference Riscal13,Reference Arena14], and higher ROS levels were associated with increased cytarabine sensitivity [Reference Hosseini15]. Cytarabine and anthracyclines are standard drugs for AML [Reference De Kouchkovsky and Abdul-Hay16], and gemcitabine has recently been suggested as drug candidate for paediatric AML [Reference Drenberg17]. This may be of clinical relevance in AML patients in whom MDM2 inhibitor treatment may modify the efficacy of next-line therapies, even if there is no response to MDM2 inhibitor therapy.
Conclusion
Our data do not provide evidence that MDM2 inhibitors may exert mutagenic effects that would promote the formation of loss-of-function TP53 mutations. MDM2 inhibitors rather seem to select TP53-mutant cells after mutations have occurred. Surprisingly, we found that cultivation of PL21 cells in the presence of ineffective nutlin-3 concentrations resulted in increased drug sensitivity in a substantial fraction of sublines. This is conceptually important, because our findings show that non-effective therapies can affect the outcome of next-line therapies. A better understanding of such processes may inform therapy protocols in the future. Our study also illustrates how cancer cell lines as permanent preclinical model systems can be used to produce findings that cannot be made in the clinics, because different treatment schedules cannot be compared in the same patient.
Author Contributions
J.C. and M. Michaelis designed and conducted the study. C.S., F.R., T.R., M. Mernberger, and A.N. performed experiments. All authors analysed data. M. Michaelis and J.C. wrote the initial manuscript draft. All authors read and approved the final version.
Funding Information
The work was supported by the Hilfe für krebskranke Kinder Frankfurt e.V. (J.C.), the Frankfurter Stiftung für krebskranke Kinder (J.C.), the Deutsche José Carreras Leukämie-Stiftung (J.C., T.S.), and the Kent Cancer Trust (M. Michaelis).
Publishing Ethics
The authors confirm that
1. the manuscript has been submitted only to the journal – it is not under consideration, accepted for publication or in press elsewhere. Manuscripts may be deposited on pre-print servers;
2. all listed authors know of and agree to the manuscript being submitted to the journal; and
3. the manuscript contains nothing that is abusive, defamatory, fraudulent, illegal, libellous, or obscene.
Conflict of Interest
The authors declare none.
Data Availability
All data are included in the manuscript.





 Open access
Open access
Comments
Comments to the Author: Michaelis and co-workers aim at the verification of the hypothesis of the generation of TP53 mutations upon the treatment of p53wt PL21 cells with MDM2 antagonist, nutlin-3. Also, increased susceptibility of nutlin-treated cells to three anti-cancer drugs is reported. While this study is important, the manuscript suffers from several critical conceptual mistakes which largely decrease its impact.