


1. Introduction
Several unsupervised Bayesian clustering approaches have been proposed to infer population genetic structure using exclusively molecular marker information obtained from sampled individuals. These methodologies can be used to determine the number of clusters (K) and to assign individuals to the inferred clusters (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Dawson & Belkhir, Reference Dawson and Belkhir2001; Corander et al., Reference Corander, Waldmann, Marttinen and Sillanpaa2004; Gao et al., Reference Gao, Williamson and Bustamante2007; Huelsenbeck & Andolfatto, Reference Huelsenbeck and Andolfatto2007). Loosely speaking, such methods (e.g. the one implemented in the program STRUCTURE) are developed based on population genetics models and try to infer population genetic structure by minimizing Hardy–Weinberg and linkage disequilibrium within the different groups.
However, related individuals or members of the same family can be present in the same subpopulation. Such a situation can occur when individuals are sampled from small populations, from populations of high fecund species, or when sessile individuals are sampled. Consequently, the lack of Hardy–Weinberg and linkage equilibrium which may arise from this situation could lead to a reduction in the accuracy to detect population structure with the program STRUCTURE (Pritchard et al., Reference Pritchard, Wen and Falush2009). To our knowledge, three studies (Guinand et al., Reference Guinand, Scribner, Page, Filcek, Main and Burnham-Curtis2006; Anderson & Dunham, Reference Anderson and Dunham2008; Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012) have investigated the effects of the presence of close relatives on the power of the software STRUCTURE (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Falush et al., Reference Falush, Stephens and Pritchard2003) to estimate K. Using real and simulated data, these studies showed that the presence of close relatives led to the false detection of population genetic structure when it is absent or to the overestimation of the number of clusters. Rodríguez-Ramilo & Wang (Reference Rodríguez-Ramilo and Wang2012) evaluated the improvement achieved by first identifying and removing all family members but one using a relatedness analysis (Wang, Reference Wang2004; Wang & Santure, Reference Wang and Santure2009) before conducting population structure analysis. This study found better performance of the clustering method implemented in the software STRUCTURE (i.e. higher accuracy in terms of estimated number of clusters) once the samples had been pruned.
Another approach to circumvent the problem of related individuals when inferring population genetic structure could be using clustering methodologies which do not make assumptions about Hardy–Weinberg and linkage equilibrium. Dupanloup et al. (Reference Dupanloup, Schneider and Excoffier2002) proposed a spatial procedure that uses a simulated annealing algorithm to find the arrangement that maximizes the proportion of total genetic variance due to differences between groups of populations. With a similar rationale, Rodríguez-Ramilo et al. (Reference Rodríguez-Ramilo, Toro and Fernández2009) developed an approach (implemented in the software CLUSTER_DIST; http://dl.dropbox.com/u/5714008/Fernandez.htm) based on the maximization of the mean genetic distance between inferred populations (also solved through a simulated annealing algorithm). This approach was developed based on the idea that highly differentiated populations show a large genetic distance between them. This distance can be calculated from the molecular marker information without assumptions on Hardy–Weinberg or linkage equilibrium. In Rodríguez-Ramilo et al. (Reference Rodríguez-Ramilo, Toro and Fernández2009) the actually implemented distance was the Nei minimum distance (Nei, Reference Nei1987), which can be calculated from the pairwise molecular co-ancestry between individuals (Caballero & Toro, Reference Caballero and Toro2002) or directly from the allelic frequencies.
In the present study, the algorithm implemented in CLUSTER_DIST was evaluated in the presence of related individuals (i.e. full-sib and half-sib families). The performance of this method, in terms of the number of estimated clusters, is compared with the performance of the software STRUCTURE using simulated data.
2. Materials and methods
(i) Simulations
The simulated data were the same as in Rodríguez-Ramilo & Wang (Reference Rodríguez-Ramilo and Wang2012). Briefly, the allele frequencies at a locus for the entire population, p i (i = 1, 2,…, L; where L is the number of genotyped loci), were drawn from a uniform Dirichlet distribution, with all parameters set to a value of 1. From these allele frequencies and the parameter F ST, the particular allele frequencies of subpopulation j, p ij (i = 1, 2,…, n; where n is the number of subpopulations), were then obtained also from a Dirichlet distribution with parameters p i (1−F ST)⁄F ST. Using the allele frequencies of each subpopulation, the genotypes of individuals from a given subpopulation were then randomly generated for each marker locus. A further generation was simulated to create families of individuals with a given relationship, with genotypes of each descendant obtained following the rules of Mendelian transmission independently for each locus. Individuals with a particular family structure (detailed below) were sampled from the 2nd generation of each subpopulation for clustering analysis.
The simulations considered scenarios involving 3 or 5 subpopulations (n) with 50 individuals per subpopulation and, thus, a total census size of 150 or 250 individuals (N); genetic differentiation level F ST = 0·1 or 0·2; individuals genotyped for 10 (20) microsatellite-like markers with 20 alleles each or 100 (200) bi-allelic markers (mimicking single nucleotide polymorphisms (SNPs)); 0, 1 or 2 sib families (in the same subpopulation or in different subpopulations) comprising 4 or 16 siblings per family; and full-sib or half-sib (only simulated for 10 microsatellites) families simulated. The combination of all these factors resulted in a huge number of different scenarios.
(ii) Algorithms
STRUCTURE (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Falush et al., Reference Falush, Stephens and Pritchard2003) was run with 5000 sweeps of burn-in and 10 000 sweeps of data collection in each replicated scenario, using the admixture and the correlated allele frequency models. All other settings were left with their default values. The range of possible K values evaluated was from two to the simulated number of subpopulations plus one (4 or 6). Note that, following Anderson and Dunham (Reference Anderson and Dunham2008) to reduce computation time, STRUCTURE was run only one time per replicated scenario, then the estimation of K could not be done using Evanno et al. (Reference Evanno, Regnaut and Goudet2005) criterion, because it needs several runs of STRUCTURE for each evaluated K.
However, other alternative criteria have been proposed to estimate K from the output of STRUCTURE. One of these alternative criteria (which does not need several runs of STRUCTURE) is to estimate K from the proportion of inferred ancestry (Q). For a given value of K, STRUCTURE provides the value
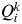 $Q_i^k $
, the proportion of the inferred ancestry of individual i (i = 1,…, N) being from cluster k (k = 1,…, K). For each individual i, the cluster that has the highest Q value is the cluster to which this individual belongs to (given a particular value of K). The average of the
$Q_i^k $
, the proportion of the inferred ancestry of individual i (i = 1,…, N) being from cluster k (k = 1,…, K). For each individual i, the cluster that has the highest Q value is the cluster to which this individual belongs to (given a particular value of K). The average of the
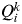 $Q_i^k $
for all individuals i belonging to cluster k is symbolized as
$Q_i^k $
for all individuals i belonging to cluster k is symbolized as
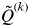 $\tilde Q^{(k)} $
. The smallest value of
$\tilde Q^{(k)} $
. The smallest value of
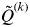 $\tilde Q^{(k)} $
across clusters is denoted as
$\tilde Q^{(k)} $
across clusters is denoted as
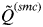 $\tilde Q^{(smc)} $
, and this value indicates to what extent individuals are assigned unambiguously to the clusters or whether they show mixed ancestry.
$\tilde Q^{(smc)} $
, and this value indicates to what extent individuals are assigned unambiguously to the clusters or whether they show mixed ancestry.
Analyses of many simulated and real data sets indicated that
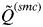 $\tilde Q^{(smc)} $
decreases slowly as the number of K increases until the true number of subpopulations is reached (see Anderson & Dunham, Reference Anderson and Dunham2008; Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012 for more details). Thereafter,
$\tilde Q^{(smc)} $
decreases slowly as the number of K increases until the true number of subpopulations is reached (see Anderson & Dunham, Reference Anderson and Dunham2008; Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012 for more details). Thereafter,
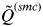 $\tilde Q^{(smc)} $
decreases rapidly with an increasing value of K. According to this, the inferred number of clusters was determined to be the highest value of K that has
$\tilde Q^{(smc)} $
decreases rapidly with an increasing value of K. According to this, the inferred number of clusters was determined to be the highest value of K that has
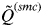 $\tilde Q^{(smc)} $
> 0·8. This threshold value depends on the number and polymorphism of the evaluated molecular markers, and showed a high efficiency in the simulations considered in this study (Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012).
$\tilde Q^{(smc)} $
> 0·8. This threshold value depends on the number and polymorphism of the evaluated molecular markers, and showed a high efficiency in the simulations considered in this study (Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012).
CLUSTER_DIST (Rodríguez-Ramilo et al., Reference Rodríguez-Ramilo, Toro and Fernández2009) is based on the idea that highly differentiated populations show a high genetic distance between them. This distance can be calculated from molecular markers without taking into account Hardy–Weinberg and linkage equilibrium assumptions. Several genetic distances have been proposed (Laval et al., Reference Laval, Sancristobal and Chevalet2002). Among those distances, the Nei minimum distance (Nei, Reference Nei1987) has the advantage that it can be calculated through the pairwise molecular co-ancestry between individuals (Caballero & Toro, Reference Caballero and Toro2002).
Notwithstanding, a shortcoming of the method is that no measure of confidence is obtained for the final arrangement of clusters. This problem is circumvented in the actual implementation of CLUSTER_DIST by using an allele frequency approach. The considered configurations, instead of assigning each individual to a single cluster, are lists of vectors (one for each individual) carrying their probability to belong to each cluster. Consequently, the sum of positions (i.e. probabilities) for a particular individual equals one. In the final (optimal) configuration those individuals with a probability close to one of belonging to a particular cluster can be assigned with great confidence. Contrarily, assignment of individuals with lower probabilities will carry a higher uncertainty, possibly reflecting the presence of admixture or the insufficient amount of information to assign this individual to a single cluster. Realize that the algorithm does not explicitly allow for admixture, but tries to put individuals in completely separated groups.
The implementation of the method within CLUSTER_DIST uses a simulated annealing algorithm to find the partition that showed the maximal average genetic distance between subpopulations. Simulated annealing is an optimization procedure adequate to deal with many genetic problems (e.g. Fernáandez & Toro, Reference Fernández and Toro1999). Parameters used to run the CLUSTER_DIST software in this study included 10 000 alternative solutions generated per step, the maximum number of steps (temperatures) is set to 250, and an initial temperature (T) of 0·00001, which was reduced for each step by a factor of Z (cooling factor) equal to 0·9. CLUSTER_DIST was also run one time per replicated scenario. For each simulated scenario, the range of possible K values evaluated with CLUSTER_DIST ranged between two and six. The number of inferred clusters was estimated with an approach following the same rationale as Evanno et al. (Reference Evanno, Regnaut and Goudet2005), but adapted to genetic distances (please see Rodríguez-Ramilo et al., Reference Rodríguez-Ramilo, Toro and Fernández2009 for more details).
(iii) Measurement of accuracy
Twenty replicates of each combination of the considered parameters were carried. Accordingly, the proportion of the 20 replicates, where the compared software identified the correct number of subpopulations (3 or 5), was used as the accuracy measure.
3. Results
Figure 1 shows the proportion of replicates where the number of clusters was correctly inferred (i.e. estimate was K = 3) by STRUCTURE and CLUSTER_DIST when the real number of subpopulations was n = 3 in the presence of full-siblings. The figure presents results for the two differentiation levels among subpopulations (F ST = 0·1 and 0·2) and the two sets of microsatellites-like markers (10 and 20).

Fig. 1. Proportion of replicates where STRUCTURE (left panels) and CLUSTER_DIST (right panels) infer K = 3 when n = 3 in the presence of full-siblings. Dashed and solid lines indicate 10 and 20 microsatellites, respectively. Triangles represent one family, squares indicate two families in the same subpopulation, and circles represent two families in different subpopulations.
The accuracy of both STRUCTURE and CLUSTER_DIST decreases as the size of the families increases. In fact, both methodologies show a high accuracy when there are no siblings. However, the deterioration in performance with the presence of sib families is less pronounced in the case of CLUSTER_DIST (right panels), especially when the differentiation between subpopulations is high (F ST = 0·2). Expectedly, the use of more markers results in a higher accuracy for both methodologies. There is not a clear effect of the number of families when the total number of full-siblings in a sample is fixed. Similar results to those observed for microsatellite loci were also obtained for full-siblings using 100 or 200 SNPs (Fig. 2).

Fig. 2. Proportion of replicates where STRUCTURE (left panels) and CLUSTER_DIST (right panels) infer K = 3 when n = 3 in the presence of full-siblings. Dashed and solid lines indicate 100 and 200 SNPs, respectively. Triangles represent one family, squares indicate two families in the same subpopulation, and circles represent two families in different subpopulations.
Figure 3 shows the proportion of replicates in which K is correctly inferred by STRUCTURE and CLUSTER_DIST when n = 3 in the presence of half-siblings and using 10 microsatellites. Results and conclusions obtained from them are in agreement with those of previous figures. However, for the same family size and number of families, full-sibs (Fig. 1) produced a greater reduction in accuracy than half-sibs (Fig. 3). In the half-sib scenario, it seems that a greater number of families do lead to a less accurate estimate of K.

Fig. 3. Proportion of replicates where STRUCTURE (left panels) and CLUSTER_DIST (right panels) infer K = 3 when n = 3 in the presence of half-siblings, using 10 microsatellites. Triangles represent one family, squares indicate two families in the same subpopulation, and circles represent two families in different subpopulations.
Results obtained for n = 5 with microsatellites, SNPs and full- or half-siblings were very similar to those showed above for three subpopulations (Figs. S1, S2 and S3 in Supplementary Material).
4. Discussion
The existence of close relatives within a subpopulation may lead the program STRUCTURE to infer a wrong number of groups as the assumptions of Hardy–Weinberg and linkage equilibrium within subpopulations will not be met. This has been already shown by some authors (Guinand et al., Reference Guinand, Scribner, Page, Filcek, Main and Burnham-Curtis2006; Anderson & Dunham, Reference Anderson and Dunham2008; Rodríguez-Ramilo & Wang, Reference Rodríguez-Ramilo and Wang2012). Families are really genetic structures, but are usually not the focus of most studies which are interested in knowing the organization at the population level. But depending on the final objective of the study, and the particular situation, families may become the relevant grouping, especially when differentiation levels (i.e. F ST) are low between subpopulations.
To avoid the bias of clustering methods when relatives are present, Rodríguez-Ramilo & Wang (Reference Rodríguez-Ramilo and Wang2012) proposed a simple 2-step alternative consisting of (1) detecting with the software COLONY (Wang, Reference Wang2004; Wang & Santure, Reference Wang and Santure2009) the confounding family structures from a sample and removing all but one of the members of each family and then (2) conducting a STRUCTURE (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Falush et al., Reference Falush, Stephens and Pritchard2003) analysis with the reduced sample. The first stage of this 2-step procedure trims the data and makes them to better meet the assumptions of the specific clustering methodology, greatly improving the accuracy of a population structure analysis with the software STRUCTURE. But the final results will be influenced by the accuracy of the estimator of relationships and to what extent the assumptions of the chosen methodology are fulfilled by the data.
In this paper, we introduced another alternative to improve the population genetic structure analyses in the presence of close relatives. This is a one-step procedure that uses a clustering method that is insensitive to deviations from Hardy–Weinberg and linkage equilibrium. Using this approach, there is no need of pre-correcting the data and, therefore, we avoid the possible bias that could appear if the data do not fit the assumptions of the method used to detect the family structure. Accordingly, the results presented in this study indicate that this one-step methodology is less influenced than STRUCTURE when dealing with samples containing close relatives. For this reason, CLUSTER_DIST can be used, in combination with the program STRUCTURE, to infer population genetic structure when close relatives are supposed to be present in a sample.
Removing relatives prior to inferring genetic structure using the genetic distances approach has a negligible effect (data not shown). This is another evidence of the little influence of the presence of relatives within subpopulations on the power of this clustering strategy.
5. Supplementary material
The online Supplementary Material can be found available at http://dx.doi.org/10.1017/S0016672314000068
We are very grateful to one anonymous referee for helpful comments on the manuscript.
Financial support
This work was funded by the Spanish government project (Consolider Ingenio Aquagenomics: CSD200700002).
Statement of Interest
None.




