Mitochondrial membrane protein-associated neurodegeneration (MPAN) is a subtype of neurodegeneration with brain iron accumulation (NBIA) caused by chromosome 19 open reading frame 12 (C19orf12) mutations.Reference Hartig, Iuso and Haack 1 Similar to pantothenate kinase-associated neurodegeneration, another form of NBIA, iron deposition in the globus pallidus and substantia nigra is increased in MPAN.Reference Schneider, Hardy and Bhatia 2 Symptoms may include gait impairment, spasticity, dystonia, optic atrophy, neuropsychiatric disturbances and usually present in childhood or early adulthood.Reference Olgiati, Doğu and Tufekcioglu 3 We hereby present a MPAN patient with myoclonus and hydrocephalus.
A 28-year-old man admitted to our outpatient clinic with complaints of memory deficit, tremor, inability to speak, generalized slowness, and severe balance problems making walking impossible. History was obtained from his family, as he was unable to speak. His memory deficit and slowness complaints started when he was 26, and tremor appeared a few months later. He had been diagnosed with Parkinson’s disease and had received levodopa treatment. He, however, did not benefit from this treatment and his symptoms progressed rapidly. He was unable to walk and speak for 2 weeks at the time of our evaluation. No perinatal conditions, childhood head trauma or central nervous system infections, which may lead to hydrocephalus, were reported. Although his family history was not significant, his parents were related. His father’s grandfather and his mother’s great-grandfather were siblings. He had four brothers and two sisters, all of whom were alive and did not have any neurological disorders. He had three uncles and one aunt from his father’s side; six uncles and three aunts from his mother’s side and none of them had similar symptoms.
Neurological examination revealed severe hypomimia and hypophonia, severe bradykinesia, severe rigidity, mild postural tremor of bilateral upper and lower extremities, and myoclonic jerks in the upper extremities. Myoclonic jerks were observed spontaneously at rest with the frequency increasing during voluntary movement. He was wheelchair bound, unresponsive to verbal cues, and did not communicate in any form.
Plasma iron and ceruloplasmin levels, and hepatic copper concentration were normal. There was generalized slowing in the electroencephalography. Electromyography (EMG) did not show any signs of peripheral neuropathy, but confirmed cortical myoclonus affecting all limbs irregularly with arrhythmic spontaneous 40–50 millisecond (ms)-long bursts at rest, with increased frequency during action (Figure 1; Supplementary Video).
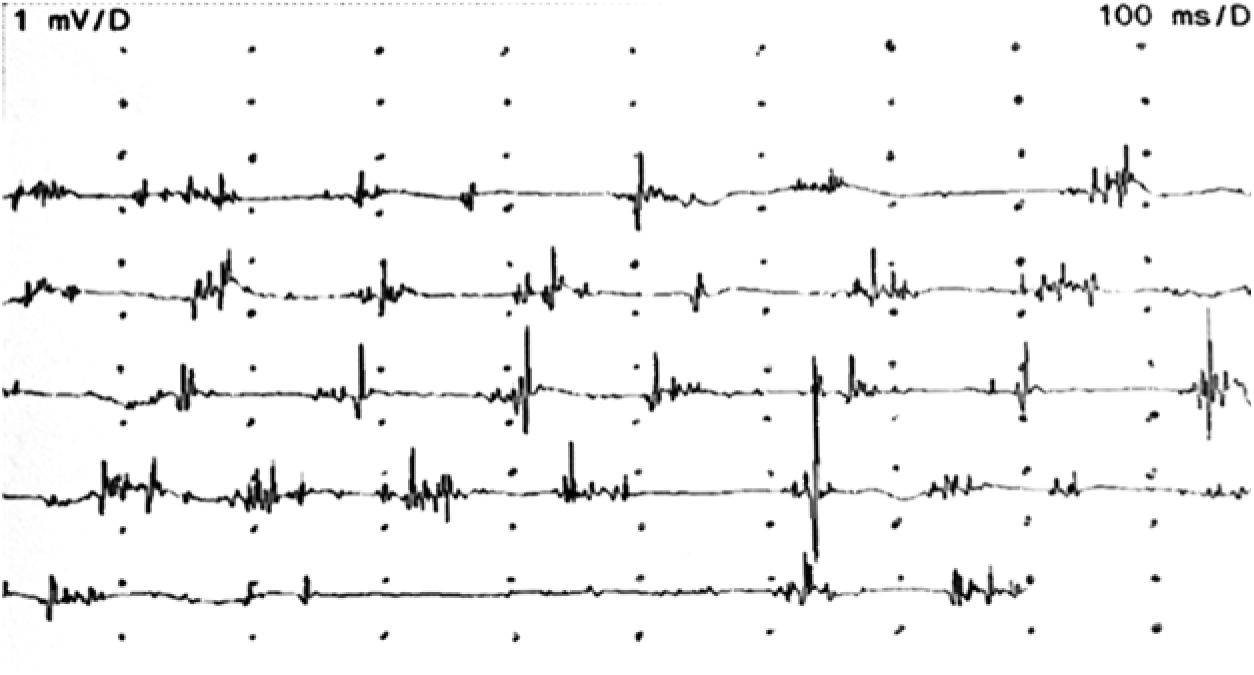
Figure 1: EMG recordings of the right flexor carpi ulnaris muscle during myoclonic jerks.
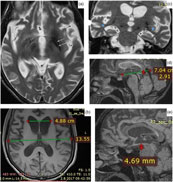
The brain magnetic resonance imaging (MRI) revealed bilateral hypointensities in globus pallidus and substantia nigra, mild brain atrophy and enlargement in temporal horns of lateral ventricles (Figure 2). Medullary lamina was prominent specifically on the left side on T2-weighted MRI. Evans index was 0.35, suggesting hydrocephalus. Mamillopontine distance was decreased, measured as 4.7 millimeters (mm). Spleniochiasmal distance was decreased, measured as −2.9 mm.
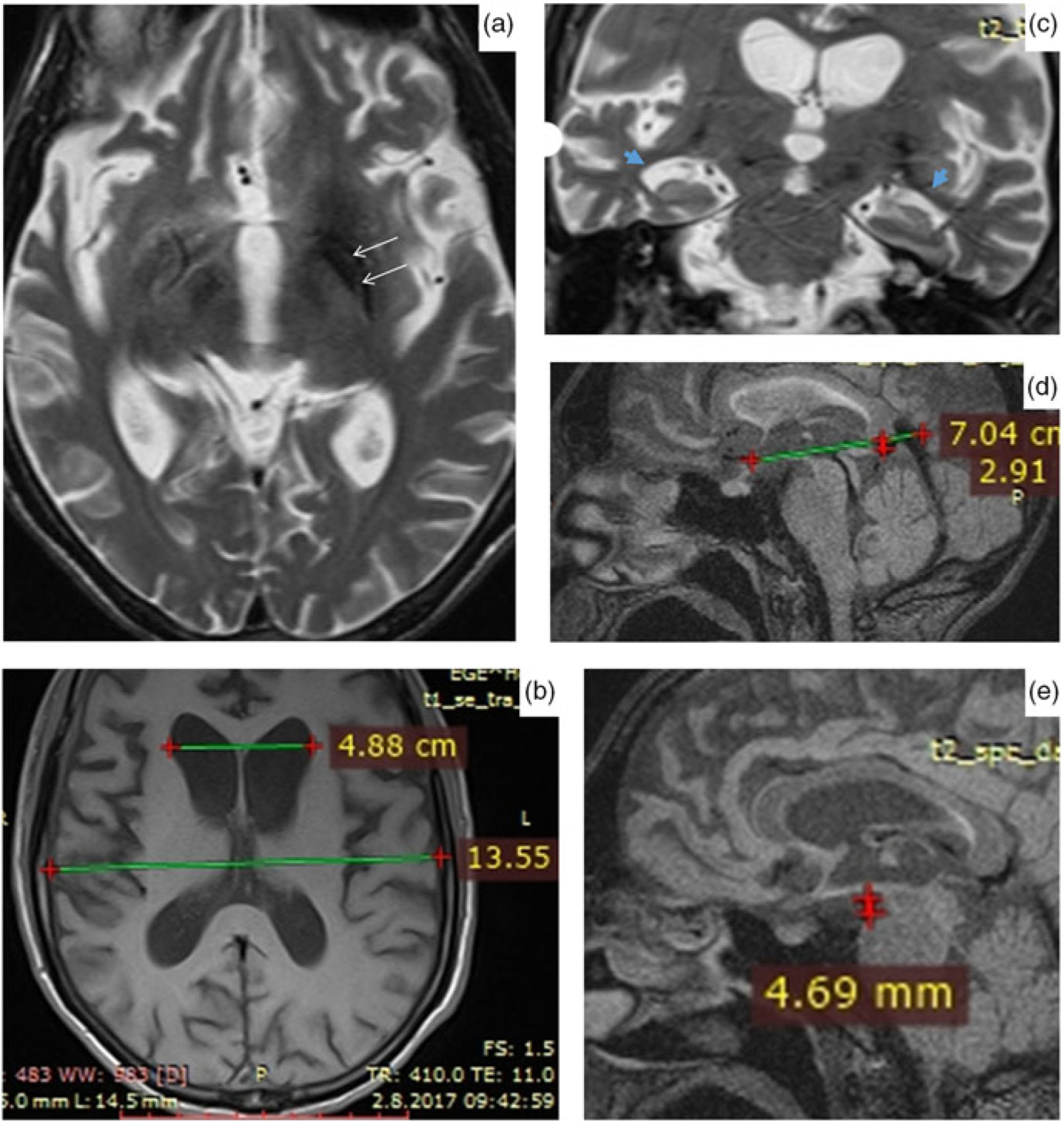
Figure 2: Magnetic resonance imaging findings. (a) Prominent medullary lamina specifically on the left side. (b) Calculation of Evan's index. (c) Enlargement in lateral ventricle temporal horns (blue arrows). (d) Calculation of mamillopontin distance. (e) Calculation of spleniochiasmal distance.
Genetic analysis revealed homozygous mutation in C19orf12 gene: c.32C > T; p. Thr11Met, leading to the diagnosis of MPAN.
Characterized by abnormal iron accumulation in the basal ganglia, NBIA is a group of disorders with a broad spectrum. MPAN has an autosomal recessive inheritance and is the third most common type of NBIA.Reference Olgiati, Doğu and Tufekcioglu 3 The disorder manifests with generalized or oromandibular dystonia, parkinsonism, spastic paralysis, and hyperreflexia in most patients. Patients are usually unable to walk by themselves by the age of 21 years.Reference Hartig, Iuso and Haack 1 Iron accumulation within basal ganglia structures leads to hypointensity in the MRI.
Our patient did not have optic atrophy, although it has been commonly reported in previous studies. Reference Hartig, Iuso and Haack1,Reference Olgiati, Doğu and Tufekcioglu3 Similarly, in another case report, optic atrophy was not observed.Reference Kłysz, Skowrońska and Kmieé 4 EMG findings also did not reveal any motor axonal neuropathy, another common finding in MPAN. Additionally, chorea in the upper extremities has been reported,Reference Kłysz, Skowrońska and Kmieé 4 whereas in our case we observed myoclonic jerks in the upper extremities which had not been reported before. The brief EMG discharges lasting less than 50 ms, irregular occurrence suggest cortical myoclonus.Reference Shibasaki 5
Hartig et al.Reference Hartig, Iuso and Haack 1 and Olgiati et al.Reference Olgiati, Doğu and Tufekcioglu 3 observed bilateral hypointensities in globus pallidus and substantia nigra on T2- or susceptibility-weighted MRI images in their case series. Olgiati et al.Reference Olgiati, Doğu and Tufekcioglu 3 reported that the hyperintensity of the medial medullary lamina between the internal and external parts of globus pallidus was also commonly observed. Additionally, we report hydrocephalus in MRI. This is a new imaging finding which has not been previously reported in MPAN. Although the patient had brain atrophy, the Evans index being over 0.30, the enlargement of the lateral ventricle temporal horns, increased frontal horn radius, increased third ventricle diameter, mild enlargement of third ventricle recesses, cingulate sulcus sign, and decreased mamillopontine diameter suggest hydrocephalus over atrophy.
Rare syndromes carry the risk of being misdiagnosed and require clear definition of their features. Thus, we hereby report that myoclonic jerks in the upper extremity and hydrocephalus in MRI can also be observed in MPAN.
Acknowledgements
The authors thank the patient and his family for their cooperation.
Funding
The authors did not receive any financial support for the preparation of this manuscript.
Disclosures
EB and EP do not have anything to disclose. MCA received honoraria from Abbott, Abdi Ibrahim, Allergan, Bohringer Ingelheim, Gen Ilaç, Generica, GlaxoSmithKline, Medtronic, Lundbeck, Novartis, Ilko, and Santa Farma. SM received her salary from CeGaT GmbH.
Statement of Authorship
EB and MCA performed the examination of the patient. EP performed the imaging data collection and analysis. SM performed the genetic analysis. All authors construed the interpretation of the analysis and contributed to the writing of the manuscript.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2019.244