


Schizophrenia is a chronic, severe and disabling psychiatric disorder that affects 1% of the population worldwide. Schizophrenia is a complex major disease that manifests as psychotic behaviour (delusions and hallucinations), disorganisation, dysfunction in normal affective responses and altered cognitive functioning. Reference van Os and Kapur1 Twin, family and adoption studies have demonstrated that schizophrenia is highly heritable; the heritability of schizophrenia is estimated to be as high as 80%. Reference Lichtenstein, Yip, Bjork, Pawitan, Cannon and Sullivan2,Reference Sullivan, Kendler and Neale3 However, the genetic loci that contribute to the disease remain generally elusive. Recent genome-wide association studies (GWAS) have provided unbiased assessments of common sequence variations across the whole genome and may robustly map the loci involved in the pathology of complex diseases. In the past 5 years, a number of GWAS of schizophrenia have been published. Reference Lencz, Morgan, Athanasiou, Dain, Reed and Kane4–Reference Shi, Levinson, Duan, Sanders, Zheng and Pe'er20 Several loci have surpassed the genome-wide significance threshold (P = 5×10−8) in more than one GWAS; for example the major histocompatibility complex (MHC) region has exhibited genome-wide significance in four independent large-scale GWAS of schizophrenia. Reference Stefansson, Ophoff, Steinberg, Andreassen, Cichon and Rujescu9,Reference Yue, Wang, Sun, Tang, Liu and Zhang14,Reference Shi, Levinson, Duan, Sanders, Zheng and Pe'er20,Reference Purcell, Wray, Stone, Visscher, O'Donovan and Sullivan21
Recently, the Schizophrenia Psychiatric GWAS Consortium (PGC) reported a large schizophrenia GWAS. 13 In this study, the authors conducted a mega-analysis of the combined genotyping data from 21 856 individuals of European ancestry from 17 separate studies and then performed a replication study in a sample of 29 839 participants from 19 populations. This study revealed seven loci with genome-wide significance, including two previously reported loci (i.e. the MHC region and TCF4) and the following five novel loci: 1p23.3 (MIR137), 2q32.3 (PCGEM1), 8p23.2 (CSMD1), 8q21.3 (MMP16), and 10q24.32–q24.33 (CNNM2/NT5C2). Moreover, a joint analysis of schizophrenia and bipolar disorder identified three additional genes that reached genome-wide significance: CACNA1C, ANK3 and ITIH3/4. The most significant new finding was the identification of MIR137, which encodes the microRNA 137, which is a known regulator of neuronal development. Notably, microRNA 137 may directly regulate some other schizophrenia susceptibility genes. Among the 301 high-confidence predicted MIR137 targets, 17 had at least one significant single nucleotide polymorphism (SNP) at P<10−4; these 17 targets included four genome-wide significant genes (i.e. TCF4, CACNA1C, CSMD1 and C10orf26). Subsequently, these four genes and ZNF804A, another compelling candidate gene for schizophrenia, were validated as MIR137 targets. Reference Kim, Parker, Williamson, McMichael, Fanous and Vladimirov22,Reference Kwon, Wang and Tsai23 These findings suggest that the MIR137-mediated pathway is involved in the aetiology of schizophrenia. In a replication study, Hamshere et al tested 78 of the 81 SNPs highlighted by the PGC in a UK population and found significant association for 37 (47%) of the SNPs. Remarkably, genetic variants in three new loci (i.e. ITIH3/4, CACNA1C and SDCCAG8) reached genome-wide significance after combining their new schizophrenia data (CLOZUK) with those of the PGC. Reference Hamshere, Walters, Smith, Richards, Green and Grozeva24 The CLOZUK sample is a series of 2640 UK individuals that were registered for clozapine treatment and had clinical diagnoses of schizophrenia and 2878 controls. Reference Hamshere, Walters, Smith, Richards, Green and Grozeva24
Only a few schizophrenia GWAS have been conducted in non-Western populations. One of these was our GWAS of the genotypes of 3750 individuals with schizophrenia and 6468 healthy controls from Han Chinese populations (BIOX GWAS). Reference Shi, Li, Xu, Wang, Li and Shen15 None of the SNPs within the 10 loci reported by PGC met the criteria for genome-wide significance in our data-set. However, the criteria for genome-wide significance minimises the occurrence of false positives (i.e. type I errors), whereas the occurrence of false negatives (i.e. type II errors) was almost certain. In another schizophrenia GWAS that was conducted in a Han Chinese population, Yue et al Reference Yue, Wang, Sun, Tang, Liu and Zhang14 identified the susceptibility locus at the MHC region. We ascertained significant associations between the MHC region, the TCF4 gene and schizophrenia in our previous study of 2496 patients with schizophrenia and 5184 normal controls drawn from a Han Chinese population, but we failed to confirm an association between the NRGN gene and schizophrenia. Reference Li, Li, Chen, Zhao, Wang and Huang25 Regarding the psychiatric traits, most of the associations were population specific; however, in some cases, the associations may exhibit convergence of risk genes (but not necessarily risk alleles) across populations. Reference Gelernter, Kranzler, Sherva, Almasy, Koesterer and Smith26 It is of great interest to replicate the findings related to the other loci that were identified by the PGC in the Han Chinese population. In the present study, we conducted a two-stage analysis. We first analysed the SNPs (n = 2595) of the eight newly identified loci, predicted the targets of microRNA 137 in the BIOX GWAS data and selected 18 candidate SNPs. These SNPs were genotyped and tested for their associations with schizophrenia in a replication cohort that consisted of 3585 patients with schizophrenia (the schizophrenia group) and 5496 controls (the control group) of Han Chinese ancestry. A meta-analysis was performed to combine the Chinese data-sets (BIOX GWAS and replication).
Method
Participants
Participants in the schizophrenia group were in-patients or out-patients who were recruited from various mental health centres. The patients were interviewed by two independent psychiatrists and were diagnosed according to DSM-IV criteria, 27 and had 2-year histories of the disorder. All met the following two criteria: preoccupation with one or more delusions and frequent auditory hallucinations. However, none of the following symptoms were prominent: disorganised speech, disorganised or catatonic behaviour, or flat or inappropriate affect. All healthy controls were randomly selected from Chinese Han volunteers (from hospitals and a community survey) who were asked to reply to a written invitation to evaluate their medical histories. Potential lists of controls were screened for suitable volunteers by excluding individuals with major mental illnesses. The sample consisted of 3585 people in the schizophrenia group (1901 men and 1684 women, the mean age at onset of schizophrenia was 35.0 years, s.d. = 11.0) and 5496 controls (2819 men and 2677 women with a mean age of 46.4 years, s.d. = 14.1). Of the participants, 1329 in the schizophrenia group and 2037 in the control group were northern Han Chinese; 1700 in the schizophrenia group and 2622 in the control group were central Han Chinese; and 556 in the schizophrenia group and 837 in the control group were southern Han Chinese. Approval was received for our study from the local Human Genetic Resources Ethics Committee of Human Genetic Resources. After providing a complete description of the study to the participants, written informed consent was obtained.
BIOX GWAS quality control
The gender established via data genotyping was checked for each of the participants, and individuals in the schizophrenia group of unknown or inconsistent gender (compared with the sample record) were removed (n = 49). Arrays with call rates <95% were excluded (n = 276). SNPs with call rates <95% in either the schizophrenia or the control group were removed (n = 92 324). SNPs with minor allele frequencies <3% (n = 228 267) and those that significantly deviated from Hardy–Weinberg Equilibrium (HWE; P≤1×10−6) among the control group (n = 28 657) were also excluded. Heterozygosity rates were calculated with the intent of removing deviations that exceeded six standard deviations from the mean; however, no samples were excluded based on this criterion. PLINK's identity by descent analysis was used to detect cryptic relatedness. When a pair of individuals exhibited a PI_HAT >0.25, the member of the pair with the lower call rate was excluded from the analysis (n = 146). Population ancestry assessments were evaluated using principal components analysis, and all of the samples were of Chinese ancestry.
SNP selection and genotyping
The allelic frequencies of the nine genome-wide significant SNPs reported in the PGC and PGC+CLOZUK studies differ widely between the European and Chinese populations (see online Table DS1, which is based on HAPMAP CEU (Utah residents with Northern and Western European ancestry from the Centre d'Etude du Polymorphisme Humain project collection) and CHB (Han Chinese in Beijing, China) data-sets), which indicates genetic heterogeneity. Therefore, we tended to select the SNPs that exhibited more significant associations in the BIOX GWAS dataset. Moreover, the linkage disequilibrium information about the chosen SNPs and those reported in the PGC and PGC+CLOZUK studies was also considered. Ultimately, 18 SNPs from 12 genes were selected for this study (online Table DS2) and further information about the selection of SNPs is provided in the Results section.
The MassARRAY iPLEX Gold platform (Sequenom, San Diego, California, USA) was used for genotyping. Polymerase chain reaction (PCR) amplification primers and single-base extension unextended primers were designed using the online software Human GenoTyping Tools (www.mysequenom.com/Tools). The genotyping analysis was performed according to the manufacturer's protocol (Sequenom). Genotype callings were extracted by the MassARRAY Typer software (version 4.0). In total 9173 individuals, including 3648 in the schizophrenia group and 5525 control groups, were genotyped. Individuals with less than 90% call rates (which may indicate poor DNA quality) were excluded from the analysis. Therefore, 9081 samples (3585 in the schizophrenia group and 5496 in the control group) were kept for further analysis. For each SNP, the call rates and P-values that resulted from HWE testing in the control group were calculated for data quality control.
Analyses
HapMap genotypes and haplotype data were downloaded using Haploview. Reference Barrett, Fry, Maller and Daly28 HWE tests, allelic association tests and meta-analysis of each SNP were conducted using PLINK. Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira and Bender29 The heterogeneity across studies was evaluated using the Q-test. The Mantel–Haenszel method was used to calculate the fixed-effect estimate. We stratified our samples into northern, central and southern groups according to their geographic region, performed analysis of each group and then combined the results via meta-analysis in the replication study.
Results
Comparison of the BIOX and PGC/PGC+CLOZUK GWAS data-sets in terms of the newly identified loci
Nine genome-wide significant SNPs within the eight loci were reported in the PGC and PGC+CLOZUK studies (PGC: 1p21.3, 2q32.3, 8p23.2, 8q21.3 and 10q24.32-q24.33, PGC+CLOZUK: ITIH3/4, CACNA1C and SDCCAG8), and their frequencies differed widely between the European and Chinese populations (Table DS1). In the PGC study, the strongest association signal was observed in locus 1p21.3 (MIR137, rs1625579, P= 1.59×10−11). rs1625529 was not genotyped in the BIOX GWAS data-set; however, a proxy SNP (rs1198588, D′ = 1 and r 2 = 1 in CHB) was present. This proxy SNP exhibited marginal significance (P = 6.91×10−2), and the direction of the effect size was consistent with the PGC report. Therefore, the rs1198588 SNP was selected in the follow-up stage. In 10q24.33 (NT5C2), the reported genome-wide significant SNP was rs11191580 (P = 1.11×10−8) in the PCG study. In the follow-up phase, we selected rs732998 (P = 9.15×10−3 in the BIOX GWAS data-set), which is in complete linkage disequilibrium with rs11191580 (D′ = 1, r 2 = 1) in the HAPMAP CHB data-set. In the locus ITIH3/4, no significant SNPs were observed in the BIOX GWAS data-set. However, rs2239547 was not genotyped in the BIOX GWAS, but the SNP was included in the follow-up phase. Within the other five loci (i.e. 8p23.2, 8q21.3, 10q24.32, SDCCAG8 and CACNA1C), the genome-wide significant SNPs reported in the PGC or PGC+CLOZUK studies were not genotyped or did not exhibit associations in the BIOX GWAS data-set; thus, we selected the other SNPs within each locus that exhibited the most significant associations (P<0.05) for replacement in the follow-up study. In the loci with multiple markers that were found to be significant in the BIOX GWAS data-set, all of the SNPs with P-values below 0.01 were selected. For each locus, at least one SNP was selected with the exception of locus 2q32.3 because no significant SNPs were observed in this locus in the BIOX GWAS data-set and because, as reported in the PGC study, rs17662626 was non-polymorphic in the HAPMAP CHB population.
Results for the 17 top-scoring MIR137 predicted target genes in the BIOX GWAS data-set
Among the 17 MIR137 predicted target genes (i.e., C10orf26, TCF4, CSMD1, CACNA1C, SLC12A2, CALN1, GRIA1, ST13, CSDC2, LUZP2, GLIS2, EPHA7, CADPS2, RGS6, TBC1D12, FAM78A and C20orf108), a total of 15 (i.e. all but CSDC2 and GLIS2) with at least one SNP were genotyped in the BIOX GWAS data-set. Within these regions, 2230 genotyped SNPs were analysed and 29 SNPs (1.30%) at CSMD, CACNA1C, CALN1, CADPS2 and RGS6 exhibited significance at the level of P<0.01. Ten tagSNPs were selected for the follow-up phase.
Results of the follow-up phase and combined analysis
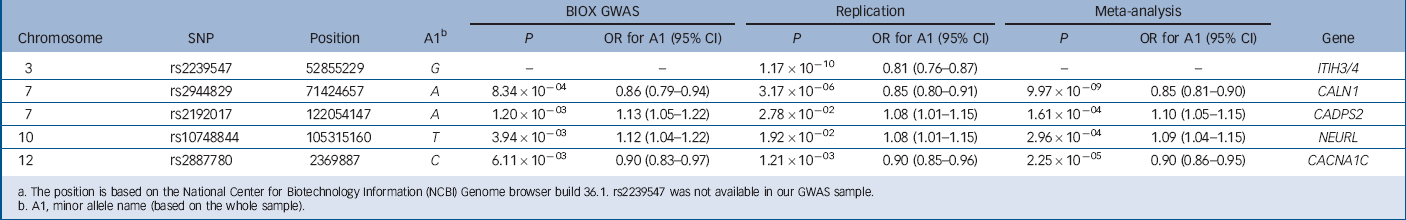
The detailed SNP quality control information is given in Table DS2, and the full results for all SNPs are listed in online Table DS3. In the follow-up study, we replicated the associations of five markers (P<0.05, with effects in the same direction, and P-values of less than 0.001 in the meta-analysis of the combined Han Chinese samples). The summary results for the five SNPs are shown in Table 1. The SNP with the most significant association was located in an intron of ITIH3/4 (rs2239547, odds ratio (OR) = 0.81, P = 1.17×10−10), which is consistent with Hamshere et al's report Reference Hamshere, Walters, Smith, Richards, Green and Grozeva24 (OR = 0.90, P = 3.62×10−10 in the PGC+CLOZUK data-set). Meta-analysis of the combined European and Chinese samples (P = 7.54×10−17, OR = 0.88; the Cochran's Q-test of heterogeneity P>0.01) strongly supported the association of this SNP.
TABLE 1 Summary of the Han Chinese population (BIOX) genome-wide association study (GWAS), replication and meta-analyses for the top five most significant single nucleotide polymorphisms (SNPs) a
| BIOX GWAS | Replication | Meta-analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | SNP | Position | A1 b | P | OR for A1 (95% CI) | P | OR for A1 (95% CI) | P | OR for A1 (95% CI) | Gene |
| 3 | rs2239547 | 52855229 | G | – | – | 1.17 × 10−10 | 0.81 (0.76–0.87) | – | – | ITIH3/4 |
| 7 | rs2944829 | 71424657 | A | 8.34 × 10−04 | 0.86 (0.79–0.94) | 3.17 × 10−06 | 0.85 (0.80–0.91) | 9.97 × 10−09 | 0.85 (0.81–0.90) | CALN1 |
| 7 | rs2192017 | 122054147 | A | 1.20 × 10−03 | 1.13 (1.05–1.22) | 2.78 × 10−02 | 1.08 (1.01–1.15) | 1.61 × 10−04 | 1.10 (1.05–1.15) | CADPS2 |
| 10 | rs10748844 | 105315160 | T | 3.94 × 10−03 | 1.12 (1.04–1.22) | 1.92 × 10−02 | 1.08 (1.01–1.15) | 2.96 × 10−04 | 1.09 (1.04–1.15) | NEURL |
| 12 | rs2887780 | 2369887 | C | 6.11 × 10−03 | 0.90 (0.83–0.97) | 1.21 × 10−03 | 0.90 (0.85–0.96) | 2.25 × 10−05 | 0.90 (0.86–0.95) | CACNA1C |
a. The position is based on the National Center for Biotechnology Information (NCBI) Genome browser build 36.1. rs2239547 was not available in our GWAS sample.
b. A1, minor allele name (based on the whole sample).
The second strongest association was observed in CALN1 (rs2944829, OR = 0.85, P = 3.17×10−6). In the combined BIOX GWAS and follow-up analysis, this locus reached genome-wide significance (P = 9.97×10−9, (A allele) OR = 0.85, 95% CI 0.81–0.90; no significant heterogeneity was observed). The PGC reported that rs12699131 (35.4 kb upstream of rs2944829 (A allele) OR = 1.10, P = 5.16×10−6) was associated with schizophrenia in the European-ancestry populations. 13 The HapMap data-set revealed that rs12699131 was in tight linkage disequilibrium with rs2944829 in the CEU+TSI (Toscani in Italia) populations (D′ = 0.94, r 2 = 0.68) and that the frequencies of the rs12699131 and rs2944829 haplotypes of the risk allele (A–G) and the protective allele (G–A) were 38.8% and 52.2% respectively. These results suggested that our finding of an association (rs2944829) is consistent with the findings (rs12699131) from individuals of European descent.
Of the eight genome-wide significant schizophrenia candidate loci identified in the European populations, the associations at ITIH3/4 (genome-wide significance) and CACNA1C (P<0.001) were replicated in our study. At locus 1p21.3, rs1198588 (MIR137) did not exhibit a significant association in the follow-up study. However, variants in three MIR137 target genes (i.e., CALN1, CADPS2 and CACNA1C) were associated with schizophrenia in the Han Chinese population at P<0.001, and the variant in CALN1 attained genome-wide significance.
Discussion
Main findings
To validate the findings of the PGC and PGC+CLOZUK schizophrenia studies (i.e. the European GWAS data-sets) in the Han Chinese population, we selected 18 SNPs from 12 schizophrenia candidate genes based on the BIOX GWAS data and then performed an association study in an extended Han Chinese replication cohort. We replicated the associations of 5/18 markers at nominal thresholds (P<0.05) with P<0.001 in the combined analysis of the BIOX GWAS and the replication, and two markers (rs2239547 at ITIH3/4, P = 1.17×10−10 and rs2944829 at CALN1, P = 9.97×10−9) reached genome-wide significance. Both of our association signals are consistent with the European findings.
Significance of our findings
Rs2239547 occurred in an extensive linkage disequilibrium block that contained many genes. Reference Hamshere, Walters, Smith, Richards, Green and Grozeva24 However, rs2239547 is an intronic SNP of ITIH4, and the expression quantitative trait loci (eQTL) data-set showed that it is significantly associated with the expression of ITIH4 (P = 3.51×10−11) in lymphoblastoid cell lines from HapMap JPT (Japanese in Tokyo, Japan) and CHB populations. Reference Stranger, Nica, Forrest, Dimas, Bird and Beazley30 Moreover, the ITIH4 protein has been found to be completely absent in patients who have had acute ischaemic strokes. Reference Kashyap, Nayak, Deshpande, Kabra, Purohit and Taori31 Moreover, patients with schizophrenia have a greater likelihood of developing stroke than do controls. Reference Tsai, Lee, Chou, Su and Chou32,Reference Lin, Hsiao, Pfeiffer, Hwang and Lee33 Thus, for this locus, ITIH4 is one of the most compelling functional candidates for further study.
We detected the first genome-wide significance of CALN1 in schizophrenia via joint analysis of the BIOX GWAS data. CALN1 has two conserved EF-hand-type calcium-binding motifs and is exclusively and highly expressed in the brain, which suggests that this gene has a role in calcium signalling in the central nervous system. Reference Wu, Lin, Liu, Jamrich and Shaffer34 Interestingly, a recent genome-wide association analysis that identified 13 new risk loci for schizophrenia suggested involvement of neuronal calcium signalling in the aetiology of schizophrenia. Reference Ripke, O'Dushlaine, Chambert, Moran, Kahler and Akterin35 Additionally, several patients with CALN1 deletions have been found to exhibit intellectual disabilities. Reference Slavotinek, Rosenfeld, Chao, Niyazov, Eswara and Bader36 Cognitive impairment is an important clinical feature of schizophrenia. We suggest that further research may clarify whether CALN1 is associated with neurocognitive phenotypes.
MIR137 and five of its targets (TCF4, CACNA1C, CSMD1, C10orf26 and ZNF804A) were reported to be associated with schizophrenia in the GWAS of individuals of European ancestry, which suggests that MIR137-mediated dysregulation is an important aetiological mechanism of schizophrenia. 13 In this study, we failed to directly replicate the association between MIR137 and schizophrenia in the Han Chinese. However, genetic variants in three predicted MIR137-target genes (i.e. CALN1, CADPS2 and CACNA1C) were significantly associated with schizophrenia; these variants included rs2944829, which reached genome-wide significance. Our findings support the hypothesis that the MIR137 pathway is involved in the aetiology of schizophrenia.
Within the eight genome-wide significant loci that were reported by the PGC or PGC+CLOZUK studies, the genome-wide significance of one locus (ITIH3/4) was replicated in our study, which provides evidence that overlapping polygenic variation exists between ethnically divergent populations. However, the other loci did not reach genome-wide significance. One possible reason for the non-replication of some genes is that the selected proxy SNPs are in extremely low linkage disequilibrium with the original genome-wide significant SNPs. These failures may also partially be explained by genetic heterogeneity across the different populations. However, other factors, such as polygenic heterogeneity, should also be considered. Moreover, the strongest signal (MIR137 locus) reported in the PGC study also failed to be replicated in the CLOZUK study. All of these phenomena highlight the genetic complexity of schizophrenia, and some susceptibility for schizophrenia is likely to result from population-specific variants. Meta-analyses and mega-analyses across different populations and larger sample sizes would efficiently aid the unravelling of this complexity. In this study, we enlarged the sample size to validate the European findings.
In summary, we independently confirmed the association of schizophrenia with ITIH3/4, which was previously reported to have genome-wide significance, and identified the genome-wide significance of CALN1, which had not previously attained genome-wide significance. Replication across different ethnic groups provides stronger evidence for the associations between schizophrenia and these two loci, and their biological mechanisms will become increasingly important for understanding of the aetiology of schizophrenia.
Funding
Design and conduct of the study: collection, management, analysis and interpretation of the data: the Natural Science Foundation, Ministry of Science and Technology and the Ministry of Education of China. Preparation, review or approval of the manuscript: Shanghai Municipal Science & Technology Commission, Education Commission and Education Development Foundation.



eLetters
No eLetters have been published for this article.