


Oxidation of LDL is accepted as an early and critical event in atherogenesis(Reference Steinberg1). Thus, high levels of oxidised LDL (oxLDL) correlate with the severity of acute coronary events(Reference Ehara, Veda and Naruko2) and are considered a biochemical marker for CHD(Reference Toshima, Haserawa and Kurabayashi3). Macrophages keep taking up oxLDL which induce the release of pro-inflammatory cytokines and promote the recruitment of monocytes and accumulation of lipid-laden macrophages named foam cells(Reference Henriksen, Mahoney and Steinberg4), which are the most predominant cell type in the earliest atherosclerotic lesions called fatty streaks(Reference Osterud and Bjorklid5). Thus, a vicious circle of oxidation, modification of lipoproteins and further inflammation can be maintained in the artery by the presence of oxLDL. In this way, the levels of circulating oxLDL have been related to intima media thickness and plaque occurrence(Reference Hulthe and Faerberg6) as well as with the progress of atherosclerosis(Reference Wallenfeldt, Fagerberg, Wikstrand and Hulthe7). The role of foam cells in the initiation and progression of atherosclerotic lesions is also extensively documented(Reference Ross8, Reference Ross9) and these cells have been shown to predispose atherosclerotic plaque to fissuring and rupture(Reference Shah, Falk, Badimon, Fernández-Ortíz, Mailhae, Villareal-Levy, Fallon, Rengstrom and Fuster10).
OxLDL cause multiple changes in cellular functions distinct from the effects of native LDL. Thus, oxLDL induce certain genes(Reference Rajavashisth, Andalibi, Territo, Berliner, Navab, Fogelman and Lusis11), suppress others(Reference Hamilton, Major and Chisolm12) and alter cellular lipid metabolism(Reference Yokode, Kita, Kikawa, Ogonochi, Narumiya and Kawai13). As well as promoting atherogenesis, oxLDL affect many aspects of macrophage function linked to the inflammatory response of these cells(Reference Berliner, Territo, Sevanian, Ramin, Kim, Bamshad, Esterson and Fogelman14). Macrophages, when activated by inflammatory stimuli, synthesise and secrete several mediators such as reactive oxygen species (ROS), cytokines, prothrombotic substances and eicosanoids, which cause, at least in part, the clinical manifestations and acute complications of atherosclerosis(Reference Libby15). Thus, it has been shown that oxLDL stimulate the release of arachidonic acid (AA) and PG by murine peritoneal macrophages(Reference Yokode, Kita, Kikawa, Ogonochi, Narumiya and Kawai13) and RAW 264.7 macrophages(Reference Akiba, Yoneda, Ohno, Nemoto and Sato16).
Diet is an important element of CVD prevention(Reference Kris-Etherton, Eckel, Howard, St Jeor and Bazzarre17). Mediterranean diets are associated with a low incidence of atherosclerotic disease(Reference Keys, Menotti and Karvonen18), but data about the specific dietary constituents involved and mechanisms conferring cardioprotection are still sparse. Olive oil is the main source of fat in the Mediterranean diet. We observed that an olive oil diet reduces AA release and subsequent metabolism through the cyclo-oxygenase-2 (COX-2) pathway in phorbol ester-stimulated macrophages(Reference Moreno, Carbonell, Sánchez, Miret and Mitjavila19). Moreover, olive oil consumption offers an additional beneficial effect by increasing NO/superoxide anion rate release by macrophages(Reference Moreno, Carbonell, Sánchez, Miret and Mitjavila19). The beneficial effects of extra-virgin olive oil have been linked to both MUFA and polyphenols(Reference Moreno and Mitjavila20). However, we must consider that other minor compounds such as β-sitosterol, α-tocopherol, terpenoids or squalene could also be contributing to these effects. Tyrosol and hydroxytyrosol are the main olive oil phenolic compounds present in extra-virgin olive oil as free or conjugated forms(Reference Owen, Mier, Giacosa, Hull, Spiegelhalder and Bartsch21) up to 800 mg/kg, and in olives (about 2 g/100 g dry weight)(Reference Marisilio, Campestre and Lanza22). These compounds show several anti-atherogenic activities, such as the inhibition of LDL oxidation(Reference Visioli, Bellomo, Nantedoro and Galli23) as well as the modulation of foam cell formation(Reference Cabello-Moruno, Perona and Ruiz-Gutierrez24). Recently, we observed that tyrosol and β-sitosterol, a phytosterol present in olive oil to appreciable concentration(Reference Parcerisa, Casals, Boatella, Codony and Rafecas25), inhibited AA mobilisation and PGE2 synthesis by macrophages stimulated by phorbol esters(Reference Moreno26). Moreover, we reported that β-sitosterol effects were the consequence of the induction of antioxidant enzymes such as glutathione peroxidase and Mn superoxide dismutase(Reference Vivancos and Moreno27).
Moderate wine consumption is another characteristic element of the Mediterranean diet. Resveratrol, present in high levels in grapes and wine (1·5–7 mg/l in red wine)(Reference Bertelli, Gozzini, Stradi, Stella and Bertelli28), has been reported to protect against atherosclerosis by modulating LDL oxidation(Reference Frankel, Waterhouse and Kinsella29), and inhibiting platelet aggregation and the production of pro-atherogenic eicosanoids by platelets and neutrophils(Reference Pace-Asciak, Rounova, Hahn, Diamandis and Goldberg30). Furthermore, we observed that the antioxidant action of resveratrol affects AA release, COX-2 induction and PGE2 synthesis induced by lipopolysaccharide or phorbol ester-stimulated macrophages(Reference Martínez and Moreno31). Recently, Delmas et al. (Reference Delmas, Jannin and Latruffe32) reported how the regulation of ROS and pro-inflammatory mediators formation by resveratrol can lead to a prevention of vascular diseases.
The aim of the present study was to determine the effect of representative minor components of wine and olive oil on ROS and eicosanoid synthesis induced by oxLDL-stimulated macrophages. Furthermore, we are interested to study the synergistic effects of these compounds.
Materials and methods
Materials and chemicals
[5,6,8,9,11,12,14,15-3H]AA (7 399 000–8 879 000 GBq (200–240 Ci)/mmol) was from American Radiolabeled Chemicals Inc. (St Louis, MO, USA). Resveratrol, β-sitosterol from soyabeans, 2,6-dichlorophenolindophenol (DCPIP), allopurinol, rotenone, fucoidan, ethidium bromide, acridine orange, catalase from human erythrocytes and superoxide dismutase from bovine erythrocytes, malondialdehyde, and 1,1′-dioctadenyl-3,3,3′,3′ tetra-methylindocarbocyanine (DiI) were obtained from Sigma Chemical Co. (St Louis, MO, USA). Tyrosol (4-hydroxyphenylethanol) was from Aldrich (Milwaukee, WI, USA). Apocynin was from Calbiochem (Darmstadt, Germany). Dulbecco's modified Eagle's medium, heat-inactivated fetal bovine serum, penicillin G, streptomycin and trypsin/EDTA were from Bio Whittaker Europe (Verviers, Belgium). All other reagents were of analytical grade.
Culture of RAW 264.7 macrophages
Murine RAW 264.7 macrophages (TIB-71) from American Type Culture Collection (Manassas, VA, USA) were grown in Dulbecco's modified Eagle's medium containing 10 % fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 μg/ml) in a 95 % air–5 % CO2 humidified atmosphere at 37°C. Cells were scraped off and passed to tissue culture in 60 mm or 100 mm dishes (Costar, Cambridge, MA, USA) for experimental purpose. Ethidium bromide–acridine orange staining was used to assess cell viability.
Isolation of low-density lipoprotein
LDL (density = 1·03–1·053) were prepared by sequential ultracentrifugation from pooled, citrated human plasma from healthy normolipidaemic volunteers, according to the method of Chung et al. (Reference Chung, Segrest, Ray, Brunzell, Hokanson, Krauss, Beaudrie and Cone33). Finally, LDL were dialysed, filtered through a 0·45 μm filter and stored at 4°C. The protein concentration was determined by the Lowry method modified by Peterson's method(Reference Peterson34).
LDL (1 mg protein/ml) were oxidised with 1·66 μm-CuSO4 at 37°C for 24 h(Reference Gieseg and Esterbauer35). Native and oxLDL were screened for lipopolysaccharide concentration by a Limulus lysate assay. All the LDL preparations used contained less than 0·75 IU lipopolysaccharide/ml. The degree of oxidation of the LDL was evaluated by a thiobarbituric acid-reactive substances (TBARS) assay as described below. Native LDL contained 0·15 (sem 0·03) nmol TBARS/mg protein, whereas oxLDL contained 51 (sem 3) nmol TBARS/mg protein. Lipoprotein preparations (LDL and oxLDL) were further dialysed against PBS containing 200 μm-EDTA at 4°C to remove free chemicals. Finally, lipoproteins were stored in the dark in sealed tubes overlaid with N2 to prevent auto-oxidation and were used within 2 weeks. The extent of LDL oxidation did not change appreciably during this period (53 (sem 3·5) nmol TBARS/mg protein, 2 weeks after oxidative induction with CuSO4).
Assay of lipid peroxides and hydrogen peroxide generation
Lipid hydroperoxide formation was measured as TBARS according to the method of Yagi(Reference Yagi36). Briefly, LDL or oxLDL (50 μg protein) were suspended in 1·5 ml of 150 mm-NaCl and mixed with 0·5 ml of 20 % TCA and 0·5 ml of thiobarbituric acid reagent (0·67 % thiobarbituric acid aqueous solution–glacial acetic acid, 1:1, v:v) and boiled at 95°C for 60 min. The mixture was cooled and shaken with 2 ml n-butanol. After centrifugation, the n-butanol layer was removed, and fluorescence was measured on a fluorescence spectrophotometer with excitation at 515 nm and emission at 550 nm. Tetramethoxypropane was used as a standard and results are expressed as nmol of malondialdehyde equivalents.
H2O2 concentration in the culture cell supernatant fraction was determined by the Mapson method(Reference Mapson37). DCPIP (1 ml, 40 μm) was reduced by ascorbic acid (20 μl, 60 mm solution), which attenuated the blue colour. H2O2 in samples (50 μl) in the presence of 5 μl of horseradish peroxidase increased absorbance (610 nm) owing to the reoxidation of DCPIP. A control reaction was performed by adding catalase. From the equimolar stoichiometry of the redox reaction involved, H2O2 concentrations were calculated using the molar extinction coefficient of oxidised DCPIP (2·1 × 104 m− 1/cm at 610 nm).
Incorporation and release of [3H]arachidonic acid and measurement of prostaglandin E2
After macrophage replication in dishes (2–3 d) and fetal bovine serum starvation (6 h), the medium was removed and replaced by 0·5 ml Dulbecco's modified Eagle's medium containing 0·1 % fatty acid-free bovine serum albumin and 0·003699 MBq (0·1 μCi) [3H]AA for 24 h. Cells were then washed three times with medium containing 0·5 % bovine serum albumin-containing medium to remove unincorporated radioactivity. After a study period, the medium was removed to determine the amount of radioactivity released and the cells overlaid with 1 % Triton X-100, and then scraped off the dishes. The amount of [3H]AA released into the medium was expressed as a percentage of cell incorporated which was determined in solubilised cells(Reference Martínez and Moreno31). The background release from untreated cells (11 (sem 2) % of [3H]AA incorporated) was subtracted from all data.
A sample of culture medium (0·25 ml) was acidified with 1 ml of 1 % formic acid. PGE2 was extracted in ethyl acetate (5 ml) and, after discarding the aqueous phase, the organic phase was evaporated under a stream of N2. PGE2 levels were determined by monoclonal enzyme immunoassay kits (Cayman Chemical Co., Ann Arbor, MI, USA) following the manufacturer's protocol.
Western blot analysis of cyclo-oxygenase-2
RAW 264.7 cells were washed twice in ice-cold PBS, scraped off into PBS containing 2 mm-EDTA and pelleted. Cell pellets were sonicated in PBS containing 2 mm-EDTA, phenylmethylsulfonylfluoride (2 μg/ml), aprotinin (20 μg/ml), leupeptin (20 μg/ml) and dimethyldithiocarbamic acid (200 μg/ml) and were separated by SDS-PAGE and blotted onto a nitrocellulose membrane using a MiniProtean II system (Bio-Rad, Hercules, CA, USA). Finally, the membranes were blocked and COX-2 was inmunodetected(Reference Martinez, Sanchez and Moreno38) using a rabbit polyclonal antiserum against COX-2 (Cayman Chemical Co.) in a 1:2000 dilution for 1 h. For β-actin immunoblotting, stripped membranes were overlaid with monoclonal anti-actin antibody (1:200) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Thus, COX-2 expression was normalised to the β-actin expression. All blots were developed using an enhanced chemiluminescence kit (Supersignal West Dura extended Duration Substrate) from Pierce (Rockford, IL, USA).
Assay for binding and/or uptake of oxidised low-density lipoprotein
OxLDL was labelled using the fluorescent probe DiI according to the methodology described by Innerarily et al. (Reference Innerarity, Pitas and Mahley39). OxLDL (1 mg protein/ml) was incubated overnight with DiI (30 μg/ml) at 37°C and, finally, DiI-labelled oxLDL was isolated by ultracentrifugation. Macrophages (1 × 106) were treated and stimulated with DiI-labelled oxLDL (10 or 50 μg protein/ml) or unlabelled oxLDL (50 μg protein/ml) for 3 h. After being washed three times with PBS, cells were lysed with 0·1 m-NaOH, and the solution was neutralised with 0·1 m-HCl. The mixture was sonicated, and the protein concentrations in the samples were adjusted to 80 μg protein/ml. Fluorescence intensity was measured with a spectrofluorometer (excitation at 524 nm and emission at 567 nm)(Reference Beppu, Watanabe, Sunohara, Ohishi, Mishima, Kawashi, Fujii and Kikugawa40).
Statistical analysis
Results are expressed as mean values with their standard errors. Differences between non-treated and treated cells were tested by Student's t test followed by the least significant difference test, as appropriate.
Results
Oxidised low-density lipoprotein-induced hydrogen peroxide production, [3H]arachidonic acid release and prostaglandin E2 synthesis by RAW 264.7 macrophages
OxLDL (40 μg protein/ml) induced time-dependent and concentration-dependent formation of H2O2, with maximum H2O2 levels reached 20–30 min after stimulation using 80–160 μg protein/ml oxLDL (Fig. 1). We observe no cell toxicity using this range of concentrations of oxLDL (data not shown). The exogenous addition of superoxide dismutase or catalase decreased H2O2 production over time despite the presence of oxLDL (Table 1). Addition of the NADPH oxidase inhibitor, apocynin (100 μm)(Reference Abid, Kachra, Spokes and Aired41), reduced the rate of H2O2 production induced by oxLDL, whereas allopurinol (100 μm) that inhibits xanthine oxidase(Reference Doehner, Schoene, Rauchhaus, Leyva-Leon, Pavitt, Reaveley, Schuler, Coats, Anker and Hambrecht42) or rotenone (5 μm), a mitochondrial respiratory chain complex inhibitor(Reference Madamanchi, Vendnov and Runge43), did not have any significant effects (Table 1).

Fig. 1 Effect of oxidised LDL (oxLDL) on H2O2 production by RAW 264.7 macrophages. (A) Time course of H2O2 determined in RAW 264.7 macrophage cultures stimulated by LDL (40 μg protein/ml; ■) or oxLDL (40 μg protein/ml; ▲). (B) Concentration–response curve of H2O2 production induced by RAW 264.7 cells stimulated with LDL (0–160 μg protein/ml; ■) or oxLDL (0–160 μg protein/ml; ▲) for 30 min. Data are the means of three experiments performed in duplicate, with standard errors represented by vertical bars. * Mean value is significantly different from that of the LDL-stimulated cells (P < 0·05).
Table 1 Effect of oxidised low-density lipoprotein (oxLDL) on hydrogen peroxide and prostaglandin E2 production by murine RAW 264.7 macrophages‡
(Mean values with their standard errors of two experiments performed in duplicate)
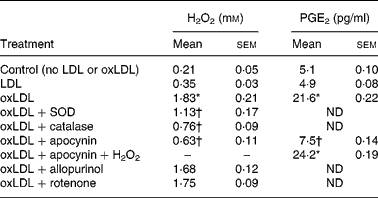
SOD, superoxide dismutase; ND, not determined.
* Mean value is significantly different from that of the LDL condition (P < 0·05).
† Mean value is significantly different from that of the oxLDL condition (P < 0·05).
‡ Cells were incubated with LDL or oxLDL (50 μg protein/ml) for 30 min at 37°C in presence of apocynin (100 μm), allopurinol (100 μm), rotenone (5 μm), SOD (50 units/ml) or catalase (1000 units/ml). H2O2 was added to 2 mm.
When [3H]AA-labelled RAW 264.7 cells were incubated with oxLDL (40 μg protein/ml), a time-dependent (Fig. 2 (A)) and concentration-dependent enhancement of [3H]AA release was observed (Fig. 2 (B)). On the other hand, macrophages incubated with native LDL showed low levels of COX-2, whereas exposure to oxLDL (40 μg protein/ml) induced an appreciable COX-2 overexpression (Fig. 3). These effects of oxLDL on AA release and COX-2 levels could be responsible to the time-dependent (Fig. 2 (C)) and concentration-dependent (Fig. 2 (D)) increase in PGE2 synthesis induced by oxLDL. Furthermore, we observed that PGE2 release induced by oxLDL was inhibited by apocynin and that H2O2 added exogenously reverted the effect of apocynin on PGE2 synthesis (Table 1). These results presented in Figs. 1 and 2 were carried out to select the conditions at which LDL and oxLDL were assayed in the rest of the study.

Fig. 2 Effect of oxidised LDL (oxLDL) on [3H]arachidonic acid (AA) mobilisation or PGE2 synthesis in RAW 264.7 macrophage cultures. (A, C) Time courses of [3H]AA release and PGE2 levels determined in RAW 264.7 macrophage cultures stimulated by LDL (40 μg protein/ml; ■) or oxLDL (40 μg protein/ml; ▲). (B, D) Concentration–response curves of [3H]AA and PGE2 release induced by RAW 264.7 cells stimulated with LDL (0–160 μg protein/ml; ■) or oxLDL (0–160 μg protein/ml; ▲) for 30 min. Data are the means of three experiments performed in duplicate, with standard errors represented by vertical bars. * Mean value is significantly different from that of the LDL-stimulated cells (P < 0·05).

Fig. 3 (A) Western blot analysis of cyclo-oxygenase-2 (COX-2) expression. Cultured RAW 264.7 macrophages were incubated with oxidised LDL (oxLDL) (50 μg protein/ml) in presence of tyrosol (10 or 100 μm, T10 and T100, respectively) or resveratrol (3 or 30 μm, R3 and R30, respectively) for 1 h. Results are representative of three separate experiments. (B) Values from (A) were normalised to the β-actin expression and are expressed as relative units with LDL expression set as 100.
Resveratrol, tyrosol and β-sitosterol modulate hydrogen peroxide production and arachidonic acid release and prostaglandin E2 synthesis induced by oxidised low-density lipoprotein
The present results show that resveratrol (30 μm) or tyrosol (100 μm) markedly decreased H2O2 levels induced by oxLDL, whereas lower concentration of resveratrol (3 μm) or tyrosol (10 μm) did not induce any significant effect (Fig. 4 (A)). However, these low concentrations of resveratrol (3 μm) together with tyrosol (10 μm) reduced significantly H2O2 production by oxLDL-stimulated RAW 264.7 cells. β-Sitosterol (10 μm) incubated in the presence of oxLDL did not modify H2O2 levels, but when the phytosterol was pre-incubated 3 h before oxLDL stimulation, we observed the impairment of H2O2 levels in macrophages. Finally, we observed that low concentrations of both polyphenols or tyrosol (10 μm) together with β-sitosterol have significant effects. Interestingly, both polyphenols and β-sitosterol reverted almost completely H2O2 production induced by oxLDL (Fig. 4 (A)).

Fig. 4 Effect of resveratrol, tyrosol and β-sitosterol on H2O2 production (A), [3H]arachidonic acid (AA) release (B) and PGE2 synthesis (C) induced by oxidised LDL (oxLDL). RAW 264.7 macrophages were incubated with resveratrol (3–30 μm) or tyrosol (10–100 μm) for 1 h or with β-sitosterol (10 μm) for 1 or 3 h, and then stimulated with oxLDL (50 μg protein/ml) for 3 h. Finally, H2O2 levels, [3H]AA release and PGE2 levels were measured. Data are the means of three experiments performed in duplicate, with standard errors represented by vertical bars. * Mean value is significantly different from that of the control (LDL) cells (P < 0·05). † Mean value is significantly different from that of the non-treated oxLDL cells (P < 0·05).
Similarly, we found that resveratrol (30 μm) and tyrosol (100 μm) significantly inhibited [3H]AA release and PGE2 synthesis (Fig. 4 (B) and (C)). Interestingly, low concentrations of resveratrol (3 μm) and tyrosol (10 μm) incubated together also impaired AA release and PGE2 synthesis induced by oxLDL. Similar effects were obtained when we used tyrosol (10 μm) together with β-sitosterol. The effect of these compounds on PGE2 synthesis could be related to the inhibition of the COX-2 overexpression induced by oxLDL (Fig. 3). Furthermore, the incubation of the low concentration of both polyphenols in the presence of β-sitosterol induced an additional appreciable inhibition of AA mobilisation and PGE2 levels induced by oxLDL (Fig. 4 (B) and (C)).
Resveratrol, tyrosol and β-sitosterol did not modify oxidised low-density lipoprotein uptake by RAW 264.7 macrophages
Finally, we examined the effects of resveratrol, tyrosol and β-sitosterol on the binding and uptake of oxLDL using DiI-labelled oxLDL. As shown in Fig. 5, incubation of RAW 264.7 cells with DiI-labelled oxLDL (10 or 50 μg protein/ml) resulted in a marked increase in fluorescence intensity, while autofluorescence of the sample, prepared from cells stimulated with unlabelled oxLDL, was less than 10 units. Under these conditions, resveratrol (30 μm), tyrosol (100 μm) and β-sitosterol (100 μm) did not modify fluorescence intensity induced by DiI-labelled oxLDL, whereas fucoidan (10 μg/ml), a polyanionic polysaccharide effective competitor for oxLDL in studies of receptor binding(Reference Krieger and Herz44), prevented the increase in fluorescence intensity.

Fig. 5 Effect of resveratrol (R), tyrosol (T) and β-sitosterol (S) on binding and/or uptake of 1,1′-dioctadenyl-3,3,3′,3′ tetramethylindocarbo-cyanine (DiI)-labelled oxidised LDL (oxLDL). Macrophages were incubated with resveratrol (30 μm), tyrosol (100 μm) or fucoidan (F; 10 μg/ml) for 1 h or β-sitosterol (100 μm) for 3 h, and then stimulated with or without DiI-labelled oxLDL (10 or 50 μg protein/ml) for 3 h. Then, the cells were washed and the fluorescence intensity in the lysate of the cells was measured. Data are the means of three experiments performed in duplicate, with standard errors represented by vertical bars. * Mean value is significantly different from that of the oxLDL-treated cells (P < 0·05). † Mean value is significantly different from that of the non-treated DiI-oxLDL cells (P < 0·05).
Discussion
Our findings show that exposure to low, non-toxic levels of oxLDL leads to the production of H2O2 by RAW 264.7 cells as was previously reported by Masella et al. (Reference Masella, Vari, D'Archivio, Di Benedetto, Matarrese, Malorni, Scazzocchio and Giovannini45). Interestingly, this effect was observed when we employed oxLDL concentrations capable of transforming macrophages into foam cells(Reference Takaku, Wada and Jinnnouchi46–Reference Rahaman, Lennon, Febbraio, Rodrez, Hazen and Silverstein48). A more detailed analysis has revealed that exogenous addition of superoxide dismutase, catalase or apocynin, a potent and selective inhibitor of the NADPH oxidase system through interfering with the translocation of an essential cytosolic protein, p47phox(Reference Abid, Kachra, Spokes and Aired41), modulate these events. This suggests that NADPH oxidase is involved in H2O2 production induced by oxLDL. Recently, similar results were reported by Rouhanizadeh et al. (Reference Rouhanizadeh, Hwang, Clempus, Marcu, Lassegue, Sevanian and Hsiai49) who observed that oxidised components of LDL induce NADPH oxidase and superoxide anion production by vascular endothelial cells.
The redox state of the cell may act as a molecular switch that regulates the activity of many enzymes and genes in concert. AA is released from cellular phospholipids by phospholipase A2 (PLA2) in response to a variety of physiological stimuli, which is the rate-limiting step in subsequent metabolism by cyclo-oxygenase pathway. We found that ROS such as H2O2 are involved in Ca2+-independent PLA2 activation and subsequent AA release in RAW 264.7 macrophages(Reference Martínez and Moreno50, Reference Martínez and Moreno51). On the other hand, we observed that ROS production could activate an early-immediate gene leading to new synthesis of COX-2 in macrophages and subsequent PGE2 synthesis(Reference Martínez, Sanchez and Moreno52). Recently, Lupo et al. (Reference Lupo, Nicotra, Giurdanella, Anfuso, Romeo, Biondi, Tirolo, Marchetti, Ragusa and Alberghina53) reported that oxLDL induced Ca2+-dependent PLA2 and Ca2+-independent PLA2 gene expression. Muroya et al. (Reference Muroya, Ihara, Ikeda, Yasuoka, Miyahara, Urata, Kondo and Kohno54) reported that oxLDL induced NFκB signalling, a redox-sensitive transcription factor involved in the regulation of COX-2 expression, in RAW 264.7 macrophages. Considering all together, the present results suggest that H2O2 production induced by oxLDL is involved in the AA release, COX-2 overexpression and PGE2 release by RAW 264.7 macrophages. Several authors have reported similar effects of oxLDL on ROS production(Reference Krieger and Herz44) or on PGE2 synthesis(Reference Yokode, Kita, Kikawa, Ogonochi, Narumiya and Kawai13, Reference Matthys, Van Hove, Jorens, Rosseneu, Marescau, Herman and Bult55). However, to our knowledge, the present study is the first to have related the effect of oxLDL on oxidative stress, AA release, COX-2 expression and the subsequent PGE2 synthesis.
In the context of the Mediterranean diet and CVD, it has also been shown that extra-virgin olive oil and wine polyphenols increase the resistance of LDL to oxidation, both in vitro and ex vivo (Reference Visioli, Bellomo, Nantedoro and Galli23, Reference Capasso, Evidente, Avolio and Solla56). Furthermore, Covas et al. (Reference Covas, Konstantinidou, Mysytaki, Fitó, Weinbrenner, de la Torre, Farre-Albadalejo and Lamuela-Raventos57) demonstrated that wine consumption reduces postprandial LDL oxidation. They also reported that olive oil ingestion reduces postprandial LDL oxidation(Reference Covas, de la Torre and Farré-Albaladejo58). Thus, although several authors reported that olive oil or wine polyphenols modulate LDL oxidation, the present results are the first to compare quantitatively the effect of minor compounds of olive oil and wine on AA cascade activation induced by oxLDL. Our findings show that olive oil and wine polyphenols such as tyrosol and resveratrol modulate H2O2 production and COX-2 overexpression induced by oxLDL as well as AA release and PGE2 synthesis. Considering that oxLDL uptake and foam cell formation take place via scavenger receptors(Reference Hiltunen and Yla-Herttuala59), and that Miles et al. (Reference Miles, Wallace and Calder60) observed that an olive oil diet decreased macrophage uptake of oxLDL though the down regulation of scavenger receptors, we attempted to explore whether polyphenols such as tyrosol and/or resveratrol influence oxLDL interaction with macrophage receptors in our experimental conditions. The present results demonstrated that the above effects were not the consequence of these compounds interfering with oxLDL binding to their receptors.
Perhaps the more interesting findings of the present study were that we observed an additive effect for the co-incubation of both polyphenols. Thus, resveratrol (3 μm) or tyrosol (10 μm) concentrations reached in plasma after wine(Reference Walle, Hsieh, DeLegge, Oatis and Walle61) or olive oil(Reference Weinbrenner, Fitó, Farré Albadalejo, Saez, Rijken, Tormos, Coolen, De La Torre and Covas62) consumption were not significantly effective on ROS and PGE2 synthesis induced by oxLDL. However, both together were markedly effective on these events under our experimental conditions. In addition, the presence of β-sitosterol, a characteristic olive oil phytosterol, enhances tyrosol and tyrosol/resveratrol actions. These synergistic effects of the phytosterol could be related to the fact that β-sitosterol activates antioxidant enzymes such as Mn superoxide dismutase and glutathione peroxidase and consequently modulates the cellular redox state(Reference Vivancos and Moreno27) though a different mechanism from tyrosol or resveratrol. Thus, it seems that the simultaneous action of polyphenols of olive oil and wine and phytosterols of olive oil could result in an additional atheroprotective effect through the modulation of the AA cascade induced by oxLDL. Most relevantly, such effects occurred at olive oil/wine minor compound concentrations within the range expected after nutritional intake from a Mediterranean diet(Reference Bertelli, Giovannini, Stradi, Bertelli and Tillement63–Reference Visioli, Galli, Bornet, Mattei, Patelli, Galli and Caruso65). Furthermore, we must consider that other phenolic compounds from olive oil such as dihydroxytyrosol, caffeic acid, kaempferol or oleuropein can raise the action of the above minor components(Reference Miles, Zoubouli and Calder66).
In conclusion, our findings suggest a new molecular mechanism by which minor components of olive oil and wine, two characteristic elements of the Mediterranean diet, may prevent the release of pro-inflammatory and pro-atherogenic AA-derived mediators by oxLDL-stimulated macrophages. Furthermore, we demonstrated an important synergistic effect of several olive oil and wine minor compounds which can explain their beneficial effects at physiological concentrations.
Acknowledgements
The present study was supported by the Spanish Ministry of Science and Technology (BFI2001-3397, BFU2004-04 960), the Spanish Ministry of Health (RD06/0045/0012) and the Autonomous Government of Catalonia (2001SGR00266). J. J. M. designed the study and prepared the paper. M. V. and J. J. M. contributed to the successful execution of the experimental work. We declare that we have no conflict of interest.






