



Introduction
Vortioxetine is a multimodal antidepressant with a unique mechanism of action.Reference Gonda, Sharma and Tarazi 1 First approved by the U.S. Food and Drug Administration for the treatment of adults with major depressive disorder (MDD) in 2013, vortioxetine has since been licensed in more than 80 countries worldwide. Clinical trials have shown vortioxetine to be effective and well tolerated for the treatment of depressive symptoms in patients with MDD across the approved therapeutic dose range of 5 to 20 mg/day.Reference Gonda, Sharma and Tarazi 1 Importantly, vortioxetine is also one of the few antidepressants with a demonstrated dose–response relationship for depression-related outcomes.Reference Thase, Mahableshwarkar, Dragheim, Loft and Vieta 2 This allows prescribers to increase the vortioxetine dose to optimize clinical response with ongoing treatment, rather than switch medication.
There are subtle, yet important, differences in the posology of vortioxetine worldwide. In the U.S., the recommended starting dose is 10 mg/day in adult patients (including those aged ≥65 years), with the prescribing information stating that the dose of vortioxetine should be increased to 20 mg/day as tolerated. 3 In the EU, the summary of product characteristics (SmPC) states that the starting and recommended dose of vortioxetine in adults aged <65 years is 10 mg/day. 4 Depending on individual patient response, the dose of vortioxetine may be increased to a maximum of 20 mg or decreased to a minimum of 5 mg once daily. In patients aged ≥65 years, the starting and recommended dose of vortioxetine is 5 mg/day. The approved product label and dosing recommendations in most other countries largely follow the EU SmPC. In 2019, vortioxetine was approved in Japan following completion of a specific local program of randomized controlled studies using fixed doses of 5, 10, and 20 mg/day. In Japan, only 10 and 20 mg/day are approved doses; 10 mg/day is the recommended starting dose, and the dose can be increased to 20 mg/day based on clinical response and tolerability. 5
Although these differences in posology may seem small, they appear to have a major impact on the prescribed doses of vortioxetine in clinical practice. In the U.S., 50% of current vortioxetine prescriptions are for the 20 mg/day dose; in comparison, this dose accounts for only 19% of vortioxetine prescriptions in the rest of the world (IQVIA, data on file). Although dosage flexibility provides the opportunity for physicians to individualize vortioxetine dosing to effectively balance management of depressive symptoms and tolerability, it is clearly important to ensure that patients receive the maximum benefit from treatment. Subtherapeutic antidepressant dosing has long been recognized to contribute to early withdrawal of treatment in patients with MDD.Reference Masand 6 Early optimized antidepressant therapy is likely to afford the best possible treatment outcomes.Reference Habert, Katzman and Oluboka 7 , Reference Oluboka, Katzman and Habert 8
The clinical development program of vortioxetine in MDD included 11 randomized, double-blind, placebo-controlled, fixed-dose, short-term studies investigating the efficacy and tolerability of the approved daily doses of 5, 10, 15, or 20 mg. Meta-analysis of change in Montgomery–Åsberg depression rating scale (MADRS) total score from baseline in these studies showed vortioxetine 5, 10, and 20 mg/day to be associated with significant reductions in the severity of depressive symptoms from baseline vs placebo, with treatment effect increasing with vortioxetine dose.Reference Thase, Mahableshwarkar, Dragheim, Loft and Vieta 2 A pooled analysis of the incidence and severity of adverse events during the course of these 11 studies found nausea and vomiting to be the most common dose-related treatment-emergent adverse events (TEAEs) associated with vortioxetine, the incidence of which plateaued at vortioxetine 15 mg/day.Reference Baldwin, Chrones and Florea 9
Following completion of the fixed-dose clinical development program in Japan, an updated analysis of the comparative efficacy and tolerability of vortioxetine 10 vs 20 mg/day in patients with MDD was warranted. Since the regulatory approvals, flexible-dose interventional studies have also been undertaken, which have allowed vortioxetine dose adjustment at the investigator’s discretion after an initial week of treatment with vortioxetine 10 mg/day. This allows for investigation in a controlled setting of clinician behavior concerning the frequency and timing of vortioxetine dose adjustment when prompted to assess clinical response (both efficacy and tolerability) from as early as 1 week after treatment initiation.
Methods
Data sources
Pooled data were analyzed from the six short-term (8-week), randomized, double-blind, placebo-controlled, fixed-dose studies conducted by Takeda/Lundbeck that included vortioxetine 20 mg/day for the treatment of MDD.Reference Boulenger, Loft and Olsen 10 –Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 15 Patients randomized to vortioxetine 20 mg/day received vortioxetine 10 mg/day for the first week, and then 20 mg/day for the remaining 7 weeks of the study period. In all studies, efficacy was assessed using the MADRS as a primary endpoint.
For the analysis of frequency and timing of dose adjustment, data were analyzed from three randomized, controlled, flexible-dose studies in which patients received vortioxetine 10 mg/day for 1 week, followed by 10 to 20 mg/day (based on the treating physician’s clinical judgment) for at least 7 weeks.Reference Montgomery, Nielsen, Poulsen and Häggström 16 –Reference Vieta, Sluth and Olsen 18 A separate analysis was also undertaken to evaluate dose-adjustment data from the open-label COMPLETE study, in which patients with MDD who experienced a partial response to monotherapy with a selective serotonin reuptake inhibitor (SSRI) or serotonin–norepinephrine reuptake inhibitor (SNRI) at an adequate dose for ≥6 weeks were switched to 8 weeks of treatment with vortioxetine 10 to 20 mg/day.Reference Fagiolini, Florea, Loft and Christensen 19
All studies included in these analyses (Supplementary Table S1) were registered at clinicaltrials.gov, conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and approved by the appropriate research ethics committees. In all studies, patients provided written informed consent for participation.
Statistical analysis
For the fixed-dose studies, efficacy analyses were performed using data from all randomized patients who received at least one dose of study medication and had at least one valid post-baseline efficacy assessment (full analysis set). For all studies, mean change from baseline in MADRS total score in each treatment arm was analyzed using a mixed model for repeated measures after 2, 4, 6, and 8 weeks of treatment. The primary endpoint for comparing vortioxetine 20 mg/day vs 10 mg/day was at week 8. The MADRS is the “gold standard” clinician-rated scale for assessing antidepressant efficacy in MDD clinical trials, and a reduction in MADRS total score of ≥2 points vs placebo is considered clinically significant.Reference Duru and Fantino 20 , Reference Montgomery and Möller 21 When comparing two active treatments, a 1-point difference in MADRS total score is considered clinically relevant.Reference Montgomery and Möller 21 , Reference Wang, Gislum, Filippov and Montgomery 22 A standard, aggregated data, random-effects meta-analysis was conducted using the estimates obtained in the individual studies. This analysis was also undertaken for individual MADRS items. Change from baseline in the Clinical Global Impression–Severity of Illness (CGI-S) total score at week 8 was also assessed.
Safety analyses were performed in the cohort of randomized patients who received at least one dose of study medication (all patients treated set). TEAEs occurring in ≥2% of patients in any study group were summarized using Medical Dictionary for Regulatory Activities preferred terms and according to time of onset: (i) over the entire 8-week treatment period; (ii) between day 1 and day 7 (ie, when patients randomized to vortioxetine 20 mg/day were receiving the starting dose of vortioxetine 10 mg/day); and (iii) from day 8 to day 56 (ie, following vortioxetine dose up-titration from 10 to 20 mg/day until the end of the 8-week treatment period). It should be noted that the incidence of adverse events is reported according to the patient’s randomized vortioxetine dosage, irrespective of the fact that all patients were treated with vortioxetine 10 mg/day during the first week of treatment.
Analysis of the timing and frequency of dose adjustment in the flexible-dose studies was performed in the all patients treated set. The proportion of patients receiving vortioxetine 20 mg/day was assessed at scheduled visits at week 1 (the earliest timepoint at which dose adjustment was permitted), week 4, and week 8.
Significance was set at P < .05. Analyses were conducted using SAS statistical software, version 9.4.
Results
Baseline characteristics
For the fixed-dose studies, safety data were available for a total of 3062 patients (982 in the placebo group and 144, 663, 298, and 975 in the vortioxetine 5, 10, 15, and 20 mg/day groups, respectively). In total, 3032 patients were included in the analysis of efficacy (full analysis set). Treatment groups were well matched at baseline in terms of sex, age, MADRS total score, and other clinical characteristics (Hamilton Anxiety Rating Scale, CGI-S, and Sheehan disability scale total scores) (Table 1).
Table 1. Summary of Demographics and Baseline Characteristics (Full Analysis Set)
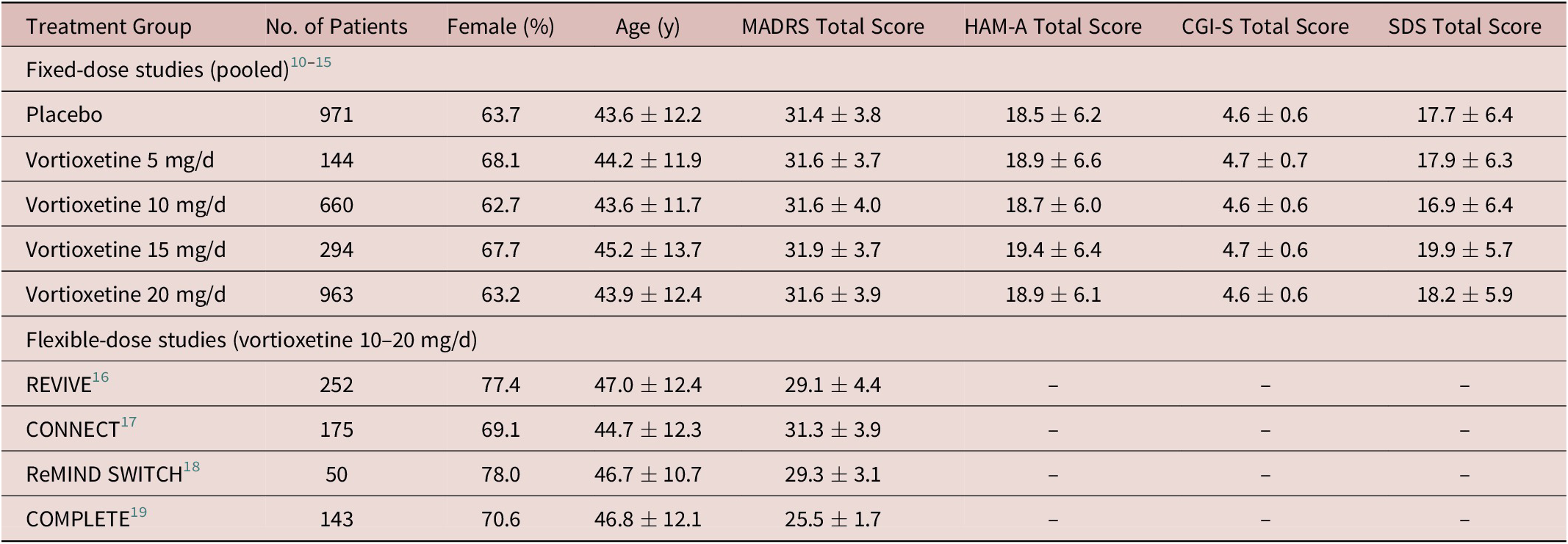
Note: All values are mean ± standard deviation unless otherwise indicated.
Abbreviations: CGI-S, Clinical Global Impression–Severity of Illness; HAM-A, Hamilton Anxiety Rating Scale; MADRS, Montgomery–Åsberg Depression Rating Scale; SDS, Sheehan Disability Scale.
Efficacy
In the fixed-dose studies, a clear dose–response relationship for vortioxetine was seen at all post-baseline timepoints assessed (Figure 1). Significant differences in mean change from baseline in MADRS total score between vortioxetine 20 mg/day and placebo were observed from week 2 onwards (P < .05 vs placebo at week 2 and P < .001 vs placebo at weeks 4, 6, and 8). A statistically significant difference vs placebo for mean change in MADRS total score from baseline after 8 weeks of treatment was seen for vortioxetine 10 mg/day (−3.11, P < .001) and 20 mg/day (−4.32, P < .001) (Table 2). Mean change in MADRS total score from baseline after 8 weeks of treatment was significantly greater for vortioxetine 20 mg/day compared with vortioxetine 10 mg/day (difference, −1.03; P < .05).
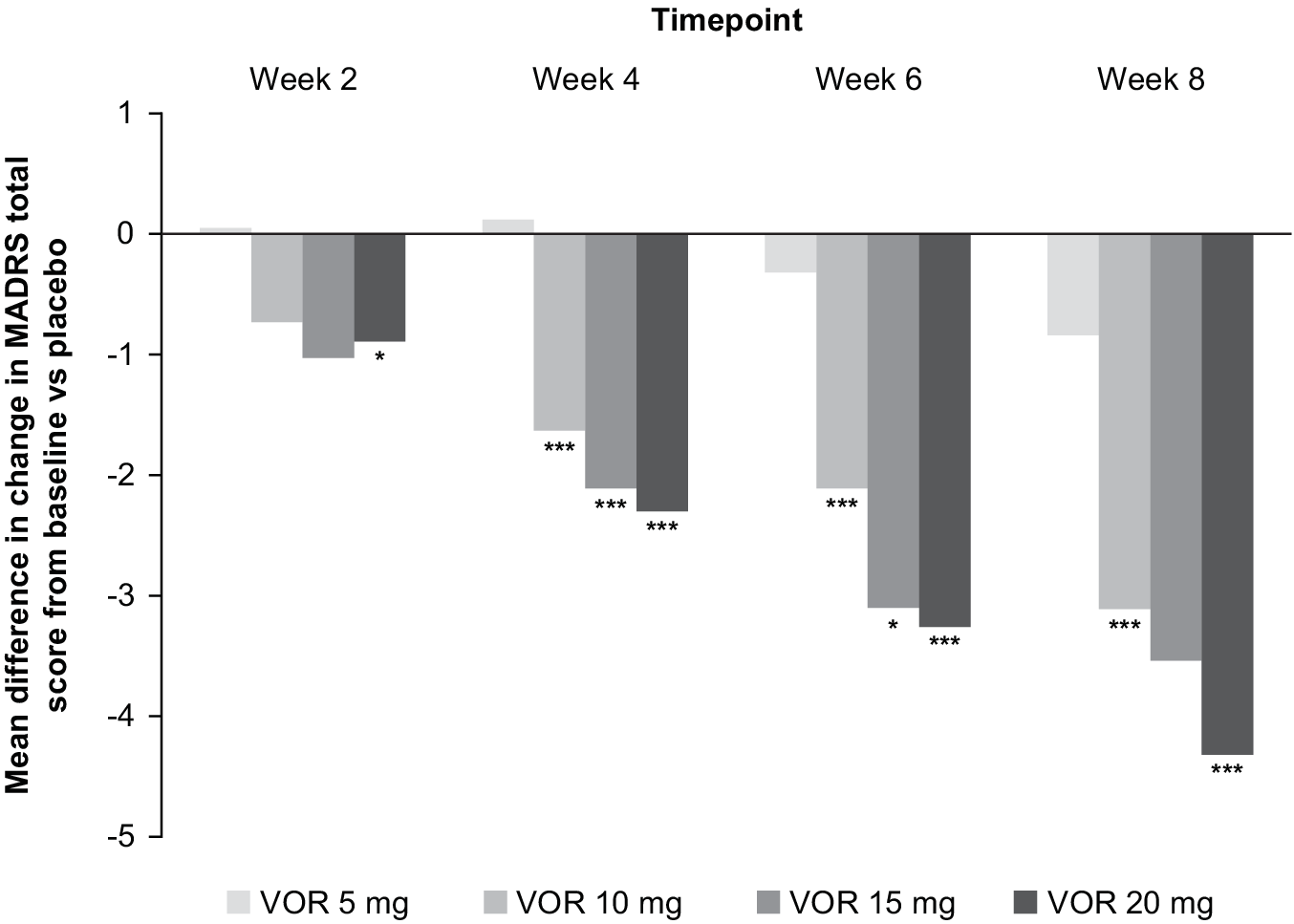
Figure 1. Mean difference in change from baseline for vortioxetine (VOR; 5, 10, 15, or 20 mg/day) vs placebo in Montgomery–Åsberg Depression Rating Scale (MADRS) total score at weeks 2, 4, 6, and 8 (full analysis set; mixed model for repeated measures analysis of six fixed-dose studies). *P < .05 vs placebo; ***P ≤ .001 vs placebo.
Table 2. Meta-Analysis of the Change from Baseline to Week 8 in MADRS Total Score Difference vs Placebo (Full Analysis Set, MMRM) in Fixed-Dose StudiesReference Boulenger, Loft and Olsen 10 –Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 15
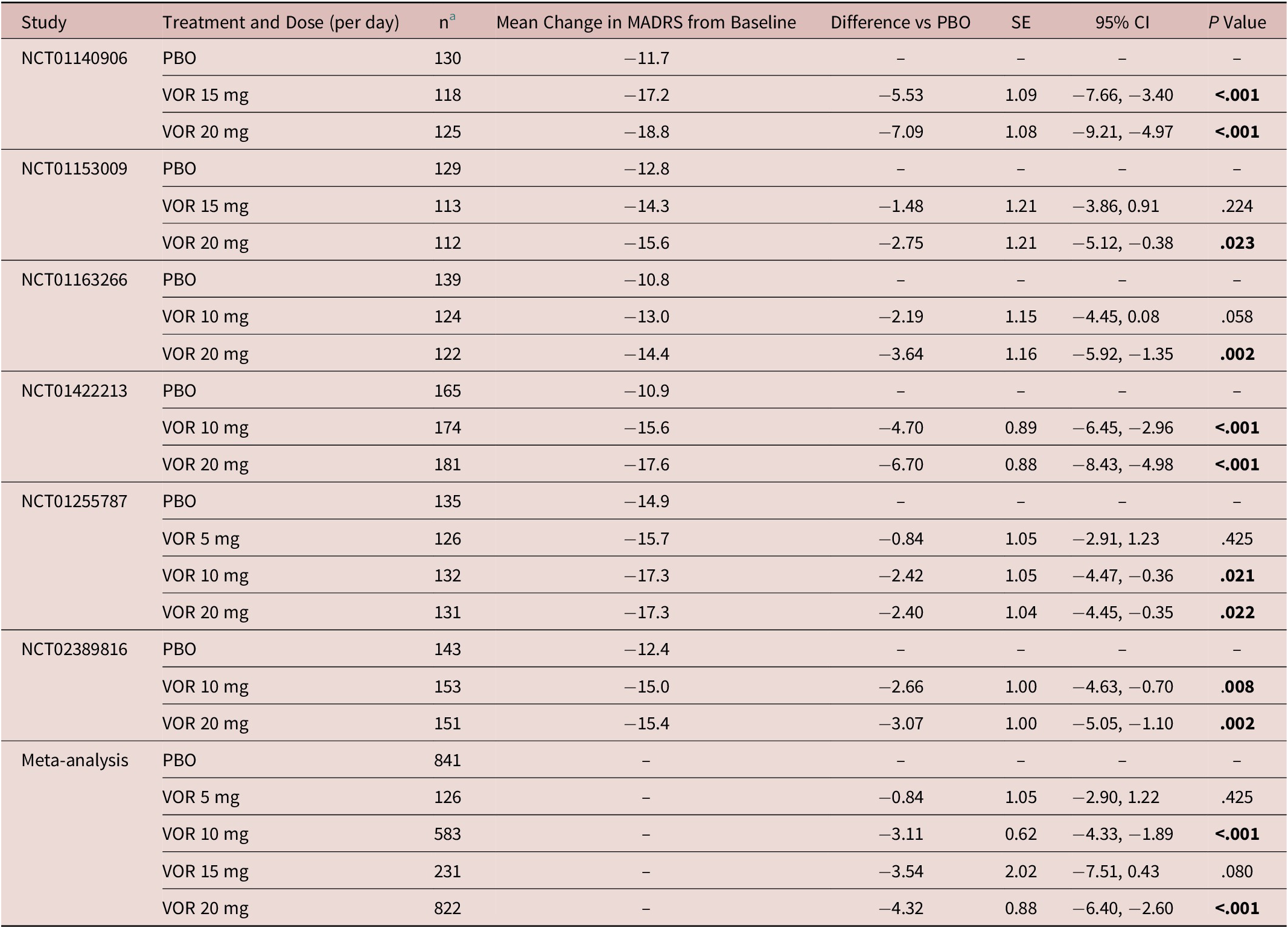
Abbreviations: CI, confidence interval; MADRS, Montgomery–Åsberg Depression Rating Scale; MMRM, mixed model for repeated measures; PBO, placebo; SE, standard error; VOR, vortioxetine.
a Number of randomized patients who received at least one dose of study medication and who had at least one post-baseline efficacy assessment (full analysis set).
A dose–response relationship was also observed for vortioxetine across all individual MADRS items at week 8 (Supplementary Figure S1). Significant differences in mean change from baseline between vortioxetine 20 mg/day and placebo were observed for the individual MADRS item scores for apparent sadness, reported sadness, inner tension, and pessimistic thoughts from week 2 onwards (Supplementary Table S2).
Similar findings were seen for CGI-S total score (Table 3). Statistically significant differences vs placebo for change in CGI-S total score from baseline after 8 weeks of treatment were shown for vortioxetine 10 mg (−0.35, P = .003) and vortioxetine 20 mg (−0.51, P < .001) (Table 3). The difference in mean change in CGI-S total score from baseline after 8 weeks of treatment for vortioxetine 20 mg compared with vortioxetine 10 mg was not statistically significant (−0.09, P = .134).
Table 3. Meta-Analysis of the Change from Baseline to Week 8 in CGI-S Total Score Difference vs Placebo (Full Analysis Set, MMRM) in Fixed-Dose StudiesReference Boulenger, Loft and Olsen 10 –Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 15
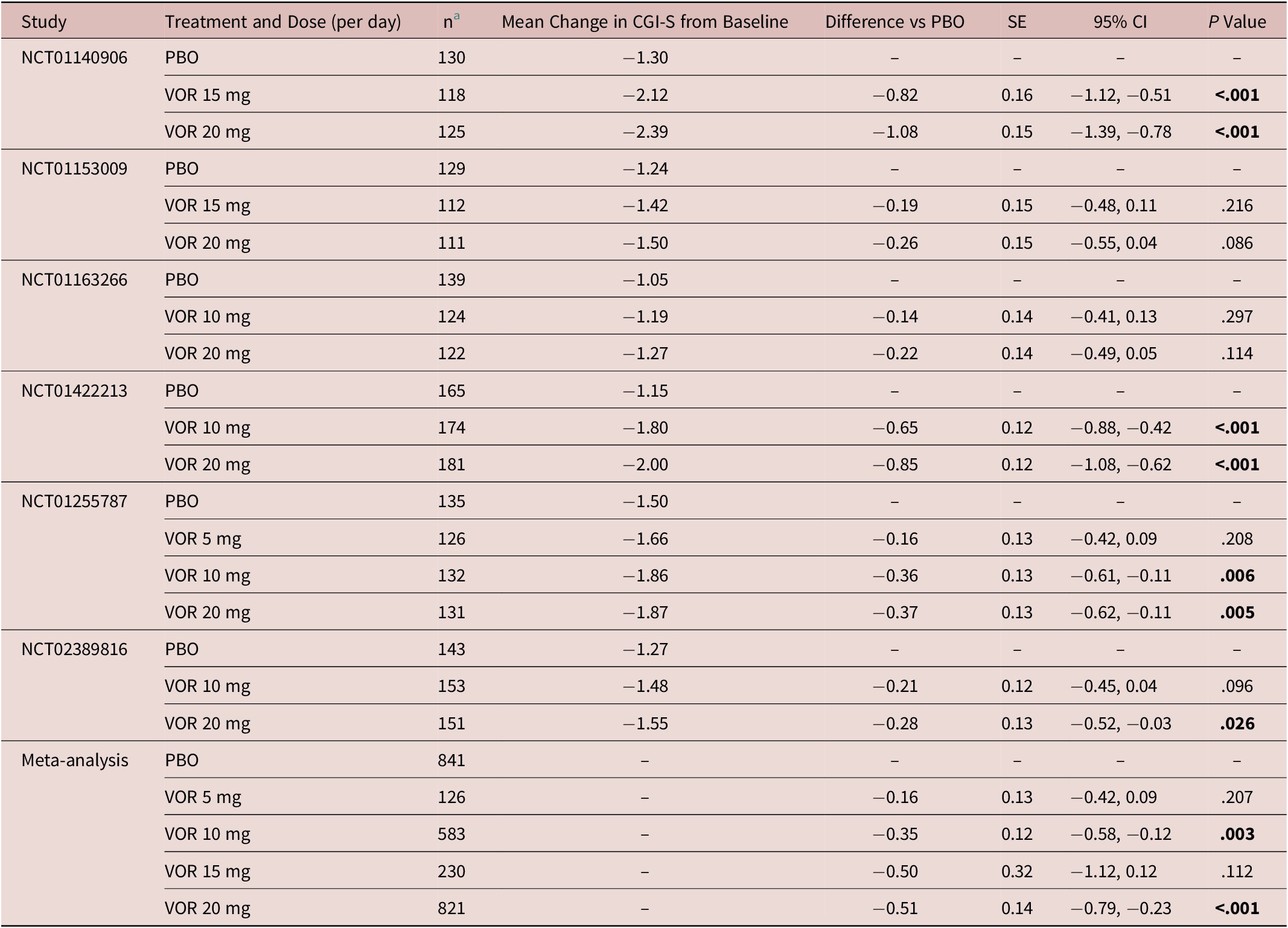
Abbreviations: CGI-S, Clinical Global Impression–Severity of Illness; CI, confidence interval; MMRM, mixed model for repeated measures; PBO, placebo; SE, standard error; VOR, vortioxetine.
a Number of randomized patients who received at least one dose of study medication and who had at least one post-baseline efficacy assessment (full analysis set).
Safety
In the fixed-dose studies, the proportion of patients reporting at least one TEAE over the 8-week, double-blind treatment period was 54.9% in the placebo group vs 66.7% in the vortioxetine 5 mg/day group, 58.1% in the vortioxetine 10 mg/day group, 66.4% in the vortioxetine 15 mg/day group, and 65.1% in the vortioxetine 20 mg/day group (Table 4). Nausea and headache were generally the most commonly reported TEAEs in vortioxetine-treated patients over the 8-week treatment period, irrespective of dose. The proportion of patients who withdrew from treatment due to TEAEs was low and similar in the vortioxetine 20 mg/day and 10 mg/day groups (6.4% and 4.4%, respectively, vs 3.3% in the placebo group). The incidence of serious adverse events was low (≤1.4% across groups), and no serious adverse event was reported by more than a single patient in any group.
Table 4. TEAEs by MedDRA Preferred Terms with Incidence ≥2% in At Least One Group in Short-Term, Randomized, Placebo-Controlled, Fixed-Dose Studies of Vortioxetine in Patients with MDD, Overall and from Day 8 (ie, Time of Vortioxetine Dose Up-Titration from 10 to 20 mg/d) Until the End of the 8-Week Treatment Period (Day 56).Reference Boulenger, Loft and Olsen 10 –Reference Inoue, Sasai, Kitagawa, Nishimura and Inada 15 All Patients in the Vortioxetine 20 mg/d Group Received Vortioxetine 10 mg/d on Days 1 to 7; Vortioxetine Dose was Up-Titrated to 20 mg/d on Day 8
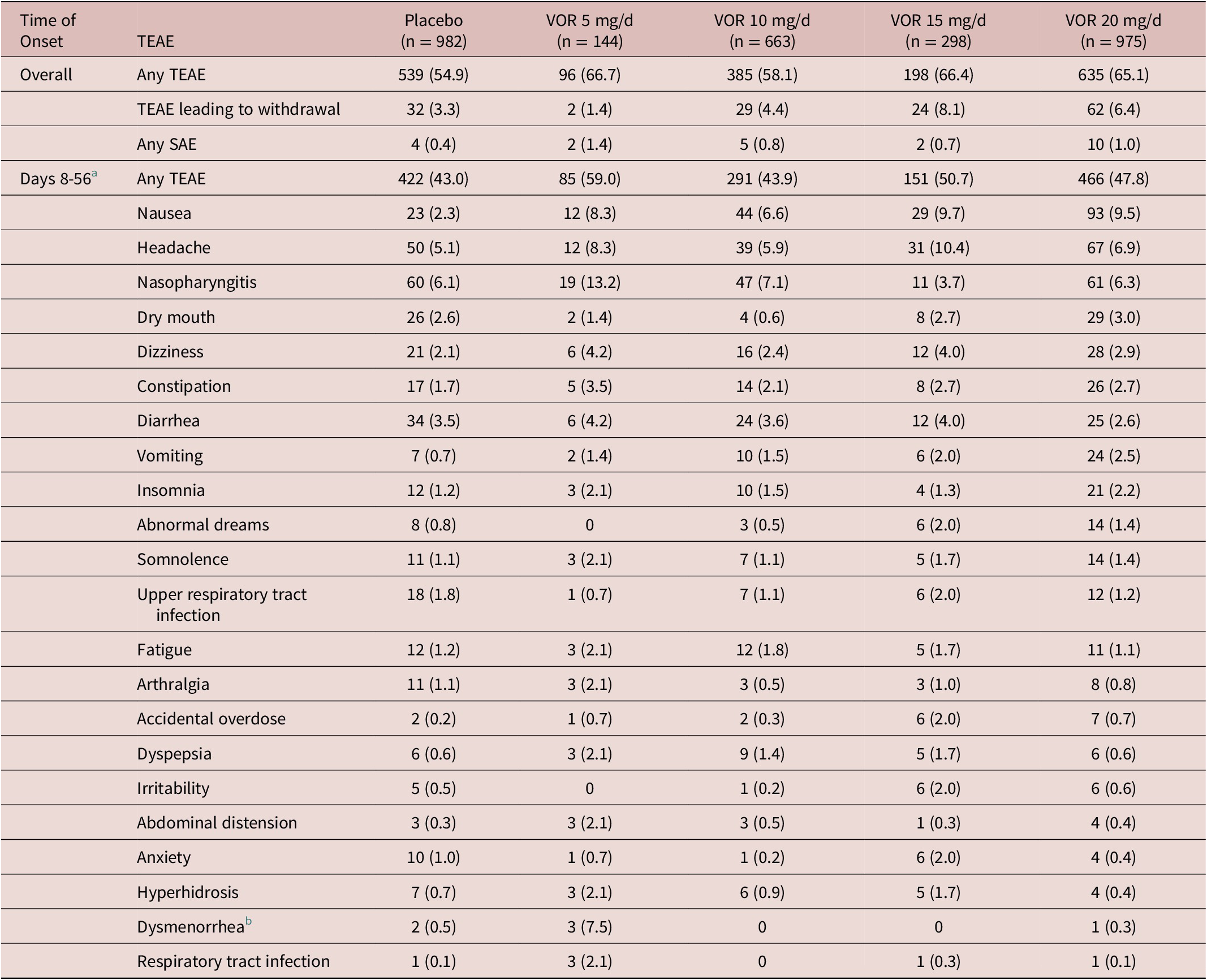
Note: All values are n (%).
Abbreviations: MDD, major depressive disorder; MedDRA, Medical Dictionary for Regulatory Activities; SAE, serious adverse event; TEAE, treatment-emergent adverse event; VOR, vortioxetine.
a Day 56, end of the 8-week treatment period.
b Sex-specific (Placebo, n = 379; VOR 5 mg/d, n = 40; VOR 10 mg/d, n = 247; VOR 15 mg/d, n = 92; VOR 20 mg/d, n = 340).
During the first week of treatment (ie, when patients in the vortioxetine 20 mg/day group were receiving vortioxetine 10 mg/day), the proportion of patients reporting at least one TEAE was 39.0% in the vortioxetine 20 mg/day group, 32.7% in the vortioxetine 10 mg/day group and 24.8% in the placebo group. Nausea and headache were the most common TEAEs with onset between days 1 and 7. The incidence of nausea during the first week of treatment was 17.9% in the vortioxetine 20 mg/day group, 13.3% in the vortioxetine 10 mg/day group, and 4.4% in the placebo group.
The proportion of patients reporting at least one TEAE from day 8 onwards (ie, following vortioxetine dose up-titration to 20 mg/day) was 47.8% in the vortioxetine 20 mg/day group, 43.9% in the vortioxetine 10 mg/day group, and 43.0% in the placebo group. Nausea, headache and nasopharyngitis were the most common TEAEs with onset between days 8 and 56 (ie, the end of the 8-week treatment period) (Table 4). The incidence of nausea with onset between days 8 and 56 was 9.5% in the vortioxetine 20 mg/day group, 6.6% in the vortioxetine 10 mg/day group, and 2.3% in the placebo group.
Dose adjustment
In the flexible-dose studies, dose increases tended to occur early after treatment initiation and subsequent reductions were uncommon. In the pooled analysis of the randomized flexible-dose studies, vortioxetine dosage was increased to 20 mg/day at week 1 in 48.0% of patients, and 64.3% were receiving vortioxetine 20 mg/day as their final dosage (at week 8 or discontinuation). Of the patients who had their vortioxetine dose increased to 20 mg/day, 9.3% subsequently had their dose decreased to 10 mg/day.
In the largest of the included randomized, controlled, flexible-dose studies,Reference Montgomery, Nielsen, Poulsen and Häggström 16 107/252 patients (42.5%) increased their vortioxetine dosage from 10 to 20 mg/day after 1 week. Of the 179 patients (71.0%) who increased their dose at any time during the study, 19 (10.6%) subsequently reduced their dose. In the open-label COMPLETE study,Reference Fagiolini, Florea, Loft and Christensen 19 vortioxetine was increased to 20 mg/day in 38/142 patients (26.8%) at week 1 (Supplementary Table S3). At week 8, 33 of these patients (86.8%) remained on vortioxetine 20 mg/day, three patients had their vortioxetine dose reduced to 10 mg/day, and two patients had withdrawn from the study. By week 8, an additional 38 patients (26.8%) had their vortioxetine dose increased from 10 to 20 mg/day. In total, 50.0% of patients were receiving vortioxetine 20 mg/day at week 8 in the study.
Discussion
This new meta-analysis of vortioxetine efficacy and tolerability not only confirms the established dose–response relationship of this antidepressant,Reference Thase, Mahableshwarkar, Dragheim, Loft and Vieta 2 but also shows vortioxetine 20 mg/day to be significantly more effective than vortioxetine 10 mg/day, with comparable tolerability. Vortioxetine was found to be well tolerated across the therapeutic dose range, with no clinically relevant increase in the incidence of TEAEs seen in patients in whom vortioxetine dosage was up-titrated from 10 to 20 mg/day after the first week of treatment. Nausea, headache, and nasopharyngitis were the most common TEAEs with onset from the time of vortioxetine dose up-titration (day 8) until the end of the 8-week treatment period (day 56), and the incidence of these events was low and similar between vortioxetine dosage groups. The proportion of patients who discontinued treatment due to TEAEs was low across all groups. No unexpected safety concerns were identified in this analysis and there was no evidence of the tolerability issues associated with other antidepressants, such as sexual dysfunction, insomnia, or weight gain.Reference Rothmore 23 –Reference Uguz, Sahingoz, Gungor, Aksoy and Askin 25
In contrast to vortioxetine 10 mg/day, vortioxetine 20 mg/day provided a clinically significant beneficial effect on depressive symptoms as assessed by MADRS total score from as early as 2 weeks. A dose-dependent response to vortioxetine was also observed across all individual MADRS items and in terms of change in CGI-S total score from baseline. These findings clearly indicate that if a patient tolerates the starting dose of 10 mg/day then the vortioxetine dosage should be increased to 20 mg/day after 1 week, in order to achieve the maximum clinical response.
When studying the frequency and timing of dose adjustment by investigators in a controlled setting, data from the flexible-dose studies showed that vortioxetine dosage was increased to 20 mg/day in almost two-thirds of all patients based on the investigators’ clinical judgment of treatment response. In most patients, this occurred early during the course of therapy, with vortioxetine dosage increased to 20 mg/day after 1 week of treatment in almost half of the patients. For patients in whom vortioxetine dosage was increased, subsequent dose reductions were uncommon. These findings are supported by those of the open-label COMPLETE study, in which 50% of patients who initially received a 10 mg/day dose were receiving vortioxetine 20 mg/day at study end.Reference Fagiolini, Florea, Loft and Christensen 19 Randomized clinical trials and interventional open-label studies can be designed to allow for earlier assessment of clinical response than may be normal in routine clinical care; in the studies included here, assessment of clinical response was mandated after the first week of vortioxetine treatment and clearly prompted a dose increase in most patients based on assessment of both efficacy and tolerability.
Collectively, our findings support increasing vortioxetine dosage to 20 mg/day early in the course of therapy in order to achieve optimal therapeutic benefit across symptoms of depression, and show that this may be achieved without compromising tolerability. In contrast to these findings for vortioxetine, a recent systematic literature review and network meta-analysis found no consistent evidence of a dose–response relationship for the antidepressant efficacy of SSRIs.Reference Braun, Adams, Rink, Bschor, Kuhr and Baethge 26 In addition, the proportion of patients withdrawing from treatment due to adverse events clearly increased with increasing SSRI dose,Reference Braun, Adams, Rink, Bschor, Kuhr and Baethge 26 suggesting that higher doses of SSRIs were less well tolerated. Indeed, for commonly used SSRIs (citalopram, escitalopram, fluoxetine, paroxetine, and sertraline), it appears that the lower range of the licensed dosages achieve the optimal balance of efficacy, tolerability, and acceptability in the acute treatment of major depression.Reference Furukawa, Cipriani, Cowen, Leucht, Egger and Salanti 27 Similarly, the SNRI venlafaxine has been reported to have an increasing dose–efficacy relationship only in the lower approved dose range (75–150 mg), with patient withdrawals due to TEAEs increasing sharply with increasing doses at this range.Reference Furukawa, Cipriani, Cowen, Leucht, Egger and Salanti 27
The above differences in dose–response and tolerability compared with other therapeutic agents for the treatment of MDD may be due to the unique pharmacologic profile of vortioxetine. Vortioxetine acts as an inhibitor of the serotonin reuptake transporter and a modulator of the activity of multiple serotonin receptor subtypes.Reference Gonda, Sharma and Tarazi 1 , Reference Sanchez, Asin and Artigas 28 , Reference Chen, Højer, Areberg and Nomikos 29 As such, vortioxetine directly and indirectly influences the activity of a range of neurotransmitter systems relevant to the pathophysiology of depression, including the serotonin, dopamine, norepinephrine, acetylcholine, histamine, glutamate, and gamma-aminobutyric acid systems.Reference Gonda, Sharma and Tarazi 1 , Reference Sanchez, Asin and Artigas 28 , Reference Chen, Højer, Areberg and Nomikos 29
Vortioxetine affects all these targets at clinically relevant doses; however, the affinity of vortioxetine varies between serotonin receptor types, resulting in dose-dependent recruitment of different mechanisms.Reference Bang-Andersen, Ruhland and Jorgensen 30 With regard to the serotonin reuptake transporter, a dose-related increase in occupancy at steady-state conditions has been observed in healthy individuals (~50% for vortioxetine 5 mg, ~65% for vortioxetine 10 mg, and >80% for vortioxetine 20 mg).Reference Areberg, Luntang-Jensen, Søgaard and Nilausen 31 It has been proposed that serotonin reuptake transporter occupancy needs to be approximately 80% to confer therapeutic benefit in MDD.Reference Meyer 32 These findings support the observed efficacy benefit of increasing vortioxetine dosage to 20 mg/day in patients with a suboptimal response to the 10 mg/day starting dose. With increasing vortioxetine dosage, there is also progressive recruitment of other serotonin receptors according to their respective affinities, which may further contribute to the overall favorable efficacy and tolerability profile of the 20 mg/day dose.Reference Sanchez, Asin and Artigas 28 , Reference Bang-Andersen, Ruhland and Jorgensen 30 , Reference Pehrson, Cremers and Bétry 33
The main limitation of our findings is that participants in clinical trials may not be fully representative of patients with MDD in routine practice settings. Furthermore, only short-term studies were included, precluding assessment of long-term dose–response effects, and patients with MDD generally require long-term treatment.
Conclusion
In summary, these findings confirm the efficacy and tolerability of vortioxetine in patients with MDD and, for the first time, demonstrate that vortioxetine 20 mg/day is both statistically and clinically more effective than vortioxetine 10 mg/day in treating the symptoms of MDD. Importantly, after the 1 week of treatment initiation with vortioxetine 10 mg/day, a similar incidence of TEAEs was observed between patients treated with vortioxetine 10 and 20 mg/day in subsequent weeks, supporting early use of the 20 mg/day dose in routine clinical practice. In randomized flexible-dose studies, in which patients received vortioxetine 10 mg/day for 1 week, after which the dose could be adjusted up to 20 mg/day based on the investigator’s clinical judgment, almost half of all patients had their dose increased to 20 mg/day after 1 week, and two-thirds of patients were receiving vortioxetine 20 mg/day as their final dosage, further supporting the efficacy, tolerability, and clinical utility of the higher vortioxetine dose.
Supplementary Materials
To view supplementary material for this article, please visit http://doi.org/10.1017/S1092852921000936.
Acknowledgments
Under the direction of the authors, medical writing support was provided by Jennifer Coward for Anthemis Consulting Ltd, funded by H. Lundbeck A/S.
Funding
The data reported in this paper were derived from clinical studies sponsored by H. Lundbeck A/S, Valby, Denmark, and Takeda Pharmaceuticals Inc., Deerfield, Illinois, USA.
Disclosures
Michael Cronquist Christensen, Ioana Florea, and Henrik Loft are employees of H. Lundbeck A/S. Andrea Fagiolini was the principal investigator for the COMPLETE study, and has been a consultant and/or speaker and/or has received research grants from Allergan, Angelini, Aspen, Boehringer Ingelheim, Daiichi Sankyo Brasil Farmacêutica, DOC Generici, FB-Health, Italfarmaco, Janssen, Lundbeck, Mylan, Otsuka, Pfizer, Recordati, Sanofi Aventis, Sunovion, and Vifor Pharma. Roger S. McIntyre has received research grant support from the Canadian Institutes of Health Research, the Chinese National Natural Research Foundation, and the Global Alliance of Chronic Diseases; has received speaker/consultation fees from AbbVie, Bausch Health, Eisai, Intra-Cellular, Janssen, Kris, Lundbeck, Neurocrine, NewBridge Pharmaceuticals, Novo Nordisk, Otsuka, Pfizer, Purdue, Sanofi, Sunovion, and Takeda; and is a CEO of Braxia Scientific Corp.
Author Contributions
All authors contributed to the conceptualization and interpretation of the analyses, contributed to the scientific content of the manuscript, and approved the final version of the manuscript for submission. H.L. analyzed the data. Employees from Lundbeck were involved in the concept of the manuscript, as well as the data analysis and interpretation of the results.








 Open access
Open access