


Despite well-established genetic diatheses and extensive research, the biochemical abnormalities underlying the predisposition to and the pathophysiology of bipolar disorder have yet to be clearly established. Early biological explanations of bipolar disorder implicated neurotransmitters, in particular the biogenic amines. However, advances in our understanding of the cellular mechanisms underlying neuronal communication have shifted the focus of research onto the role of post-receptor sites. Indeed, the ‘molecular medicine revolution’ has increased our understanding of the pathophysiological basis of a variety of medical disorders. This remarkable progress is largely attributable to the elucidation of the basic mechanisms of signal transduction, and the application of the powerful tools of molecular biology to the study of human disease. Hundreds of guanine-nucleotide-binding (G) protein-coupled receptors and over a dozen G proteins and effectors have now been characterised at the molecular and cellular level (Reference SpiegelSpiegel, 1998). This has allowed the study of a variety of human diseases which are caused by loss- and gain-of-function mutations; studies of such diseases offer unique insights into the physiological and pathophysiological functioning of many cellular transmembrane signalling pathways.
BACKGROUND
The role of G protein-coupled signal transduction pathways in human disease
G proteins are a ubiquitous family of proteins that serve the critical role of transducers of information across the plasma membrane, coupling receptors to various effectors. It has been estimated that about 80% of all known hormones, neurotransmitters and neuromodulators elicit cellular responses through G proteins coupled to a variety of intracellular effectors.
Given their widespread and crucial roles in the regulation of physiological functions, it is not surprising that alterations in G protein-coupled signal transduction pathways have been implicated in a variety of pathophysiological states: the interested reader is referred to Spiegel et al (Reference Spiegel, Weinstein and Shenker1993), Shenker (Reference Shenker1995) and Spiegel (Reference Spiegel1998) for excellent reviews. Loss- and gain-of-function mutations in G protein-coupled receptors have been identified in such diverse conditions as retinitis pigmentosa, nephrogenic diabetes insipidus, familial adrenocorticotrophic hormone (ACTH) resistance, congenital bleeding, Hirschsprung's disease, hypergonadotrophic ovarian failure, central hypothyroidism, growth hormone deficiency and familial male precocious puberty (reviewed in Reference ShenkerShenker, 1995; Reference SpiegelSpiegel, 1998). Furthermore, a variety of clinical conditions have been demonstrated to arise from alterations in the functioning of G protein subunits (Table 1) (Reference Weinstein and ShenkerWeinstein & Shenker, 1993; Reference Weinstein, Speigel, Jones and SimmondsWeinstein, 1994a ,Reference Weinstein, Spiegel, Jones and Simmonds b ). It should be noted that G protein dysfunction appears to be the primary initiating event in Albright's hereditary osteodystrophy, McCune—Albright syndrome and some endocrine tumors. In several other conditions (including hyper- and hypothyroidism, and states associated with altered glucocorticoid or gonadal steroid levels), the G protein abnormalities are probably secondary, but none the less are implicated in the pathophysiology of the condition (Reference ManjiManji, 1992; Reference Weinstein and ShenkerWeinstein & Shenker, 1993; Reference Weinstein, Speigel, Jones and SimmondsWeinstein, 1994a ,Reference Weinstein, Spiegel, Jones and Simmonds b ). For example, hypothyroidism is associated with increased levels of the inhibitory G protein (Gi) and decreased levels of the stimulatory G protein (Gs) in most tissues examined, including brain (Reference ManjiManji, 1992; Reference Weinstein, Speigel, Jones and SimmondsWeinstein, 1994a ,Reference Weinstein, Spiegel, Jones and Simmonds b ). These biochemical changes are thought to be responsible (at least in part) for the impaired lipolysis and thermogenesis observed in hypothyroidism, and may also have a role in the cognitive/psychological symptoms frequently observed. Indeed, the major effects that many hormones (including thyroid, glucocorticoids and gonadal steroids) have on signalling pathways suggest that these biochemical effects may mediate some of the observed clinical features of bipolar disorder (e.g. onset in puberty, triggering of episodes in the post-partum period, association with alterations in endogenous thyroid hormone levels and response to exogenous glucocorticoids).
Table 1 Abnormalities of signalling molecules associated with clinical conditions

| Pseudohypoparathyroidism | Gαs |
| McCune-Albright syndrome | Gαs |
| Acromegaly | Gαs |
| Hyperfunctional thyroid nodules | Gαs |
| Ovarian and adrenocortical tumours | Gαi2 |
| Congenital stationary night blindness | Gαi1 |
| Right ventricular outflow tachycardia | Gαi2 |
| Animal model of ulcerative colitis | Gαi2 |
| Hypertension | Gβ |
| Drosophila learning and memory | Adenylate cyclase |
| PKC | |
| CREB | |
| Heart failure | β ARK |
| Drosophila retinal degeneration | Arrestin |
| Amphetamine administration | RGS |
| Circadian arrhythmicity | Transcription factors |
The G protein-coupled signalling pathways first amplify and ‘weight’ extracellularly generated neuronal signals and then transmit these integrated signals to effectors, thereby forming the basis for a complex information processing network (Reference RossRoss, 1989; Reference ManjiManji, 1992; Reference Bourne and NicollBourne & Nicoll, 1993). Since a single receptor subtype can be coupled to a number of G proteins, and several G proteins can converge to activate or inactivate a single effector (Reference RossRoss, 1989; Reference TaylorTaylor, 1990; Reference ManjiManji, 1992; Reference Bourne and NicollBourne & Nicoll, 1993), these signalling pathways form complex networks. These networks facilitate the integration of signals across multiple time-scales, the generation of distinct outputs depending on input strength and duration, and regulate feed-forward and feedback loops (Bhalla & Lyengar, 1999; Reference Weng, Bhalla and LyengarWeng et al, 1999). The complexity generated by the interactions of G protein-coupled receptors may be one mechanism by which neurons acquire the flexibility for generating the wide range of responses observed in the nervous system. This has led to the proposal that G protein-coupled signal transduction pathways may be critically involved in the neuronal circuits regulating such diverse vegetative functions as mood, appetite and wakefulness and, by extrapolation, in the cellular mechanisms of action of mood-stabilising agents.
In contrast to the progress that has been made in elucidating many other medical conditions, we have yet to identify the specific abnormal genes or proteins in bipolar disorders. Genetic linkage studies suggesting the involvement of specific genes and gene products in bipolar disorder have elicited considerable excitement about the possibilities for improved treatment of this condition (Reference Gurling, Smyth and KalsiGurling et al, 1995; Gelerntner, 1995; Reference Berrettini, Ferraro and ChoiBerrettini et al, 1997; Nurnberger & Berrettini, 1998; Reference BarondesBarondes, 1998). Like other common and complex diseases such as diabetes (Reference Davies, Kawaguchi and BennettDavies et al, 1994; Reference Altman and ToddAitman & Todd, 1995) and hypertension (Reference Thibonnier and SchorkThibonnier & Schork, 1995; Reference O'Connor, Kailasam and ParmerO'Connor et al, 1996), the transmission of bipolar disorder appears to be multi-factorial in nature rather than the result of simple Mendelian inheritance (discussed in Reference Philbert, Egeland and PaulPhilbert et al, 1997). A recent study has even provided evidence for a locus for mental health wellness, on chromosome 4p at D4S2949, in pedigrees exhibiting high rates of bipolar disorder (Reference Ginns, St Jean and PhilibertGinns et al, 1998). These data suggest that the bipolar disorder phenotype may arise from a complex interplay of not only genetic and environmental risk factors, but also genetic and environmental protective factors. Moreover, differences in diagnostic criteria and aetiological heterogeneity may account for the failure to replicate many older linkage studies in bipolar disorder. More recently, several independent research groups have reported linkage of bipolar disorder to disparate chromosomal regions — 4p (Reference Blackwood, He and MorrisBlackwood et al, 1996), 6p, 13q and 15q (Reference Ginns, Ott and EgelandGinns et al, 1996) and 18q (Reference Freimer, Reus and EscamillaFreimer et al, 1996). An accompanying commentary by Risch & Botstein (Reference Risch and Botstein1996) attributed much of the uneven findings to the complex genetic mechanisms underlying the disease. Linkage studies will undoubtedly continue to be an important means of exploring the aetiology and potentially the selective treatment responsiveness of bipolar disorder; nevertheless, there is a growing consensus that other methods are needed to identify the biochemical pathways underlying the pathophysiology of what is arguably the most complex of all neuropsychiatric disorders. Indeed, it is becoming increasingly apparent that a true understanding of the pathophysiology of bipolar disorder must address its neurobiology at different physiological levels — molecular, cellular, systemic and behavioural (Reference Manji and LenoxManji & Lenox, 2000). Abnormalities in gene expression undoubtedly underlie the neurobiology of the disorder at the molecular level, and this will become evident from identification of the susceptibility and protective genes for bipolar disorder in the coming years. This must be followed by the even more difficult task of examining the impact of the faulty expression of these gene products (proteins) on integrated cell function. It is at these levels that recent studies have identified certain proteins as potential targets for the actions of mood-stabilising drugs.
There are, however, several impediments to the elucidation of the molecular and cellular mechanisms of action of mood stabilisers. First, no suitable experimental model of bipolar disorder is available, and many studies are of necessity conducted in normal rodents. Animal models of drug dependence have probably been instrumental in the pace of research on their molecular mechanisms (Reference Hyman and NestlerHyman & Nestler, 1996). Second, a problem in the identification of therapeutically relevant target genes for the actions of mood stabilisers is the lack of easily detectable phenotypic changes induced by these agents (Reference Ikonomov and ManjiIkonomov & Manji, 1999). This makes the task of ascribing functional significance to the multiple treatment-induced changes at the genomic level daunting. Moreover, establishing the genetic basis of mood as a quantitative trait is still at its inception (Reference Flint and CorleyFlint & Corley, 1996) and we therefore cannot focus on a group of already known genes. Finally, there is the dearth of knowledge concerning the neuronal circuits and pathways involved in the pathophysiology of what is probably a group of complex, heterogeneous disorders with overlapping symptom clusters, subsumed under the rubric of ‘manic—depressive illness’ or ‘bipolar disorder’.
Despite these formidable obstacles, progress is being made, using advanced cellular and molecular biological strategies to identify changes in signalling pathways and gene expression that may have therapeutic relevance in the long-term treatment of bipolar disorder. This article looks at recent research on the molecular mechanisms underlying the therapeutic effects of mood stabilisers. For discussion of some of the other signalling pathways that have been implicated in the pathophysiology and treatment of bipolar disorder, readers are referred to reviews by Wang et al, (Reference Wang, Young, Lip, Joffe and Young1997); Jope (Reference Jope1999); Li et al (Reference Li, Andreopoulos, Warsh and Reith2000); Post et al (Reference Post, Weiss, Clark, Manji, Bowden and Belmaker2000) and Warsh et al (Reference Warsh, Young, Li, Manji, Bowden and Belmaker2000).
LITHIUM AS A MOOD STABILISER IN THE TREATMENT OF BIPOLAR DISORDER
Our research laboratory has been extensively involved in the identification of specific proteins and regulatory processes involved in the action of mood-stabilising agents, particularly the monovalent cation that has had a profound effect on the lives of millions — lithium (Reference Goodwin and JamisonGoodwin & Jamison, 1990; Reference Baldessarini and TondoBaldessarini & Tondo, 2000). The discovery of lithium's efficacy as a mood-stabilising agent revolutionised the treatment of patients with bipolar disorder — indeed, it has reshaped not only medical and scientific ideas, but also popular concepts of severe mental illness (Reference Manji, Chen, Hsiao, Manji, Bowden and BelmakerManji et al, 2000a ). After nearly three decades of use in North America, lithium continues to be the mainstay of treatment for bipolar disorder, both for the acute manic phase and as prophylaxis for recurrent manic and depressive episodes (Reference Goodwin and JamisonGoodwin & Jamison, 1990; Reference Baldessarini, Tondo and HennenBaldessarini et al, 1997; Reference Davis, Janicak and HoganDavis et al, 1999; Reference Baldessarini and TondoBaldessarini & Tondo, 2000). Numerous placebo-controlled studies have strongly suggested the efficacy of lithium in the long-term prophylactic treatment of bipolar disorder, and the beneficial effects appear to involve a reduction in the number of episodes as well as in their intensity (reviewed in Reference Goodwin and JamisonGoodwin & Jamison, 1990; Reference Davis, Janicak and HoganDavis et al, 1999; Reference Baldessarini and TondoBaldessarini & Tondo, 2000). There is compelling evidence that adequate lithium treatment, particularly when given in the context of a specialist clinic, also reduces the excessive mortality observed in the illness (Reference Muller-Oerlinghausen, Ahrens and GrofMuller-Oerlinghausen et al, 1992; Reference Baldessarini, Tondo and HennenBaldessarini et al, 1997; Reference NilssonNilsson, 1999).
Despite lithium's role as one of psychiatry's most important treatments, the cellular and molecular basis for its beneficial effects have yet to be elucidated (Reference Bunney, Garland-Bunney and MeltzerBunney & Garland-Bunney, 1987; Reference Jope and WilliamsJope & Williams, 1994; Reference Manji, Chen, Hsiao, Manji, Bowden and BelmakerManji et al, 2000a ). Although a number of acute effects of lithium have been identified in vitro, its therapeutic effects in the treatment of bipolar disorder are only seen after chronic administration, thereby precluding any simple mechanistic interpretations based on its acute biochemical effects. The search for the mechanisms of action of mood-stabilising agents has been facilitated by a growing appreciation that, rather than any single neurotransmitter system being responsible for depression or mania, many interacting and overlapping systems are involved in regulating mood, and the most effective drugs are unlikely to work on any particular neurotransmitter system in isolation but, rather, affect the functional balance between interacting systems. Signal transduction pathways are therefore an attractive target to explain lithium's efficacy in treating multiple aspects of bipolar disorder (Reference Jope and WilliamsJope & Williams, 1994; Reference Manji, Chen, Hsiao, Manji, Bowden and BelmakerManji et al, 2000a ), and consequently recent research into the effects of mood-stabilising agents has focused upon second-messenger generating systems and gene expression (Reference Mork, Geisler and HollundMork et al, 1992; Reference Jope and WilliamsJope & Williams, 1994; Reference Wang, Young, Lip, Joffe and YoungWang et al, 1997; Reference JopeJope, 1999; Manji et al, Reference Manji, Moore and Chen1999, Reference Manji, Chen, Hsiao, Manji, Bowden and Belmaker2000a ; Reference Li, Andreopoulos, Warsh and ReithLi et al, 2000; Reference Warsh, Young, Li, Manji, Bowden and BelmakerWarsh et al, 2000).
Lithium and the phosphoinositide cycle: is the inositol depletion hypothesis a valid model?
In the 1990s research on the cellular mechanisms underlying lithium's therapeutic effects focused extensively upon receptor-coupled hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2). Although inositol phospholipids are relatively minor components of cell membranes, they have a major role in receptor-mediated signal transduction pathways, and are involved in a diverse range of responses in the central nervous system (reviewed in Reference Baraban, Worley and SynderBaraban et al, 1989; Reference Berridge and IrvineBerridge & Irvine, 1989; Reference Catt and BallaCatt & Balla, 1989; Reference ChuangChuang, 1989; Reference Rana and HokinRana & Hokin, 1990; Reference Fisher, Heacock and AgrenoffFisher et al, 1992). Activation of a variety of neurotransmitter receptor subtypes (including muscarinic M1, M3 and M5, noradrenergic α 1, serotonergic 5-HT2 and several metabotropic glutamatergic receptors) induces the hydrolysis of membrane phospholipids. In brief, agonists such as acetylcholine, noradrenalin (noradrenalin), serotonin or glutamate bind to specific cell surface receptors, which interact with G proteins to stimulate phospholipase Cβ (PLCβ). Activated PLC catalyses the conversion of PIP2 to two second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG): IP3 stimulates the mobilisation of intracellular calcium ions, while DAG activates protein kinase C (PKC). The trisphosphate can be phosphorylated and dephosphorylated, leading to other inositol phosphate compounds or to unphosphorylated inositol. Inositol is then converted to phosphatidylinositol (PI), which in turn is phosphorylated to phosphatidylinositol phosphate (PIP) and PIP2 (Fig. 1).
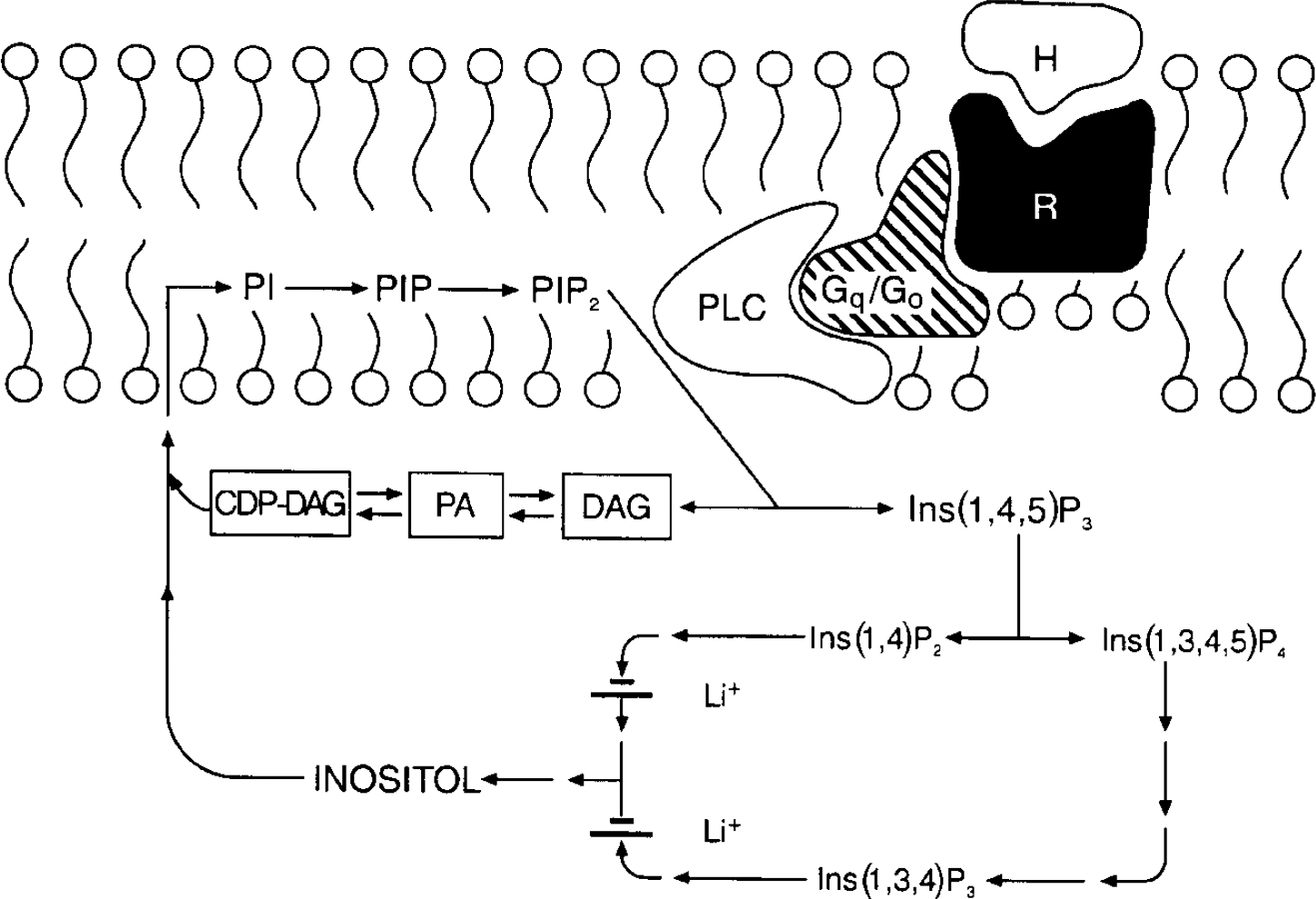
Fig. 1 The phosphoinositide cycle. The binding of a hormone or agonist (H) to receptor (R) activates G proteins which stimulate phospholipase C (PLC); PLC hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) to the two second messengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (Ins(1,4,5)P3). Ins(1,4,5)P3 stimulates the mobilisation of calcium [Ca2+]il and DAG activates protein kinase C (PKC). Ins(1,4,5)P3 can be further phosphorylated to Ins(1,3,4,5)P4 or dephosphorylated to Ins(1,4)P2. Subsequent dephosphorylations recycle inositol phosphates to inositol. Two phosphatases are inhibited uncompetitively by lithium, inositol polyphosphate-1-phosphatase (which dephosphorylates Ins(1,3,4)P3 and Ins(1,4)P2 to Ins(3,4)P2 and Ins(4)P), and inositol monophosphatase (which dephosphorylates inositol monophosphates Ins(1)P, Ins(3)P, and Ins(4)P to inositol). Free inositol (along with cytidine monophosphate phosphatidate (CMP-PA)) is essential for the synthesis of phosphatidylinositol (PI) and PIP2; CDP, cytidine 5′-diphosphate.
The ability of a cell to maintain sufficient supplies of myo-inositol (mI) is crucial to the resynthesis of the phosphoinositides, and the maintenance and efficacy of signalling. Lithium, at therapeutically relevant concentrations, is an inhibitor of inositol monophosphatase (K i 0.8 mmol/L), and results in an accumulation of inositol 1-monophosphate as well as a reduction in free inositol (Reference Allison and StewartAllison & Stewart, 1971; Reference Hallcher and ShermanHallcher & Sherman, 1980). Lithium also inhibits inositol polyphosphate-1-phosphatase, which is involved in recycling inositol polyphosphates to inositol. Furthermore, since the mode of enzyme inhibition is uncompetitive, lithium's effects have been postulated to be most pronounced in systems undergoing the highest rate of PIP2 hydrolysis (see reviews by Nahorski et al, Reference Nahorski, Ragan and Challiss1991, Reference Nahorski, Jenkinson and Challiss1992). Thus, Berridge and associates first proposed that the physiological consequence of lithium's action is derived through a depletion of free inositol, and that its selectivity could be attributed to its preferential action (due to the uncompetitive nature of the inhibition) on the most overactive receptor-mediated neuronal systems (Berridge et al, Reference Berridge, Downes and Hanley1982, Reference Berridge, Downes and Hanley1989). However, numerous studies have examined the effects of lithium on receptor-mediated PI responses, and although some report a reduction in agonist-stimulated PIP2 hydrolysis in rat brain slices following acute or chronic lithium administration, these findings have often been small and inconsistent, and subject to numerous methodological differences — see Jope & Williams (Reference Jope and Williams1994) for an excellent review. Additionally, several lines of evidence suggest that the action of chronic lithium treatment may not simply be directly manifested in receptor-mediated PI turnover. While investigators have observed that the levels of inositol in brain remain reduced in rats receiving chronic lithium, it has been difficult to demonstrate that this results in a reduction in the resynthesis of PIP2, which is the substrate for agonist-induced PI turnover (reviewed in Reference Jope and WilliamsJope & Williams, 1994).
A series of studies have recently been undertaken to determine if lithium does indeed reduce the levels of mI in critical brain regions of individuals with bipolar disorder (despite the attractiveness of the inositol depletion hypothesis, it has never been demonstrated to occur in human brain), and if so, whether these reductions are associated with its therapeutic effects.
Proton magnetic resonance spectroscopy (MRS) spectra were acquired from 8cc regions of interest (ROIs) in the frontal, temporal, parietal and occipital lobes, with an acquisition time of 5 min/ROI (STEAM pulse sequence TE 30 ms, TM 13.7 ms, TR 2000 ms (Reference Frahm, Merboldt and HanickeFrahm et al, 1987) Fig. 2). Using proton MRS, we have been able to quantitate regional brain mI concentrations with excellent reliability (Reference Moore, Bebchuk and ParrishMoore et al, 1999). Using a test—retest design (scan interval range 1-12 weeks) in six healthy volunteer subjects, the brain mI concentration (expressed in units of mI × 104/brain water and reported as the mean (standard error)) was 2.57 (0.28) v. 2.50 (0.24) with a mean difference of 0.07 or ±3% of the mI measure, demonstrating that the temporal stability and test—retest reliability of this measure are remarkably good. In order to evaluate the interrater reliability of this method, two trained individuals analysed the in vivo magnetic resonance data with MRUI-VARPRO time domain spectral analysis software (Reference Van den Boogaart, Ala-Korpela and JokisaariVan den Boogaart et al, 1994; Reference de Beer, van den Boogaart and van Ormondtde Beer et al, 1992). The individuals were masked to the study information and to each other's results. Intraclass correlation coefficient analysis revealed an interrater reliability of more than 98%.
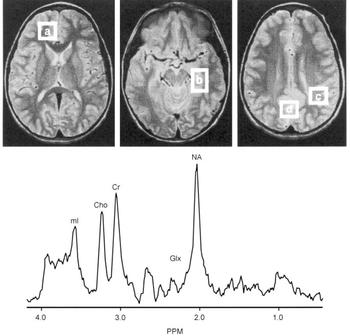
Fig. 2 Brain regions examined and typical proton magnetic resonance spectrum (MRS). Regions of interest: (a) frontal lobe, (b) temporal lobe, (c) parietal lobe, (d) occipital lobe. Lower panes: frontal lobe proton MRS from a bipolar disorder patient. PPM, parts per million, ml, myo-inositol; cho, choline compounds; Cr, creatine compounds; Glx, glutamate/glutamine/GABA; NA, N-acetyl compounds.
After extensive validation for in vivo measurement of regional brain mI concentration as well as careful determination that the 1.5 T mI resonance is indeed predominately mI (≥80%) (Reference Moore, Bebchuk and ParrishMoore et al, 1999), this method has been applied in studies of bipolar disorder. Following medication wash-out (≥2 weeks), MRS scans were performed in patients at three time points: baseline; after 5 days of lithium treatment; and after 4 weeks of lithium treatment. It was found that therapeutic administration of lithium did indeed produce significant reductions in mI levels in brain regions that had previously been implicated in the pathophysiology of bipolar disorder (Reference Moore, Bebchuk and ParrishMoore et al, 1999). However, the major lithium-induced mI reductions are observed after only 5 days of lithium administration, at a time when the patient's clinical state is completely unchanged.
Consequently, although lithium does reduce mI levels (Reference Jope and WilliamsJope & Williams, 1994), this reduction per se is not associated with a therapeutic response. Although the inositol depletion hypothesis as originally articulated does not receive support from this longitudinal human study, it remains an attractive working hypothesis that some of the initial actions of lithium may occur with the relative depletion of mI (Reference Godfrey, McClue and WhiteGodfrey et al, 1989; Reference Pontzer and CrewsPontzer & Crews, 1990; Kofman & Beimaker, 1990, 1993; Reference Tricklebank, Singh and JacksonTricklebank et al, 1991). This relative depletion of mI may initiate a cascade of secondary changes at different levels of the signal transduction process and gene expression in the central nervous system, effects that are ultimately responsible for lithium's therapeutic efficacy (Reference Manji and LenoxManji & Lenox, 1999). Studies are in progress to determine if the lithium-induced reductions in mI levels are associated with components of ultimate therapeutic response.
Lithium, valproate and the PKC signalling cascade: implications for the development of novel drugs for bipolar disorder
The PKC signalling pathway is also a target for the actions of chronic lithium (reviewed in Reference Hahn and FriedmanHahn & Friedman, 1999; Reference JopeJope, 1999; Manji & Lenox, Reference Manji and Lenox1999, Reference Manji and Lenox2000). PKC is highly enriched in brain, and plays a major role in regulating pre- and post-synaptic aspects of neurotransmission (Reference HuangHuang, 1989; Reference Stabel and ParkerStabel & Parker, 1991; Reference NishizukaNishizuka, 1992). It is now known to exist as a family of closely related sub-species, has a heterogeneous distribution in brain (with particularly high levels in presynaptic nerve terminals) and plays a major part in the regulation of neuronal excitability, neurotransmitter release and long-term alterations in gene expression and plasticity (Reference HuangHuang, 1989; Reference Stabel and ParkerStabel & Parker, 1991; Reference NishizukaNishizuka, 1992). Evidence accumulating from various laboratories has demonstrated that lithium exerts significant effects on PKC in a number of cell systems including the brain (reviewed in Reference Hahn and FriedmanHahn & Friedman, 1999; Reference JopeJope, 1999; Manji & Lenox, Reference Manji and Lenox1999, Reference Manji and Lenox2000). Most of the currently available data suggest that chronic lithium exposure results in an attenuation of phorbol ester-mediated responses, which may be accompanied by a downregulation of PKC isozymes in the brain (reviewed in Reference Hahn and FriedmanHahn & Friedman, 1999; Reference JopeJope, 1999; Manji & Lenox, Reference Manji and Lenox1999, Reference Manji and Lenox2000). Using quantitative autoradiographic techniques, it has been demonstrated that chronic lithium administration results in a significant decrease in membrane-associated PKC in several hippocampal structures. This is accompanied by isozyme-specific decreases in PKC-α and -ϵ (which have been particularly implicated in facilitating neurotransmitter release), in the absence of significant alterations in PKC-β, PKC-γ, PKC-δ or PKC-ζ (Manji et al, Reference Manji, Etcheberrigaray and Chen1993, Reference Manji, Chen, Hsiao, Manji, Bowden and Belmaker2000a ). Concomitant studies carried out in immortalised hippocampal cells in culture exposed to chronic lithium show a similar reduction in the expression of both the PKC-α and -ϵ isozymes in the cell as determined by immunoblot (Reference Manji and LenoxManji & Lenox, 1999). Chronic lithium has also been demonstrated to dramatically reduce the hippocampal levels of a major PKC substrate myristoylated alanine-rich C kinase substrate (MARCKS), which has been implicated in regulating long-term neuroplastic events (Reference Lenox, Watson and EllisLenox et al, 1992). Do these effects of lithium on PKC isozymes have any clinical relevance? Given the key role of PKC isozymes in the regulation of neuronal excitability and in neurotransmitter release, the possibility that inhibition of these isozymes represents the biochemical effect most therapeutically relevant for lithium's antimanic effects is a heuristic and testable hypothesis (discussed in more detail below). Although these effects of lithium on PKC isozmes and MARCKS are striking, a major problem inherent in neuropharmacological research is the difficulty in attributing therapeutic relevance to any observed biochemical finding. It is thus noteworthy that the structurally dissimilar antimanic agent, valproate, produces very similar effects to those of lithium on PKC-α and -ϵ isozymes and MARCKS protein (Reference Chen, Manji and HawverChen et al, 1994; Reference Watson, Watterson and LenoxWatson et al, 1998; Reference Manji and LenoxManji & Lenox, 1999; Reference Manji, Chen, Hsiao, Manji, Bowden and BelmakerManji et al, 2000a ). Interestingly, lithium and valproate appear to act on the PKC signalling pathway by different mechanisms (Reference Manji and LenoxManji & Lenox, 1999); these biochemical observations are consistent with the clinical observations that some patients show a preferential response to one or other of the agents, and that additive therapeutic effects are often observed when the two agents are co-administered (Reference Freeman and StollFreeman & Stoll, 1998).
Are PKC inhibitors effective in the treatment of acute mania?
To date, only a few studies have directly examined PCK in bipolar disorder (Reference Hahn and FriedmanHahn & Friedman, 1999). Friedman and associates investigated PCK activity and PKC translocation in response to serotonin in platelets obtained from patients with bipolar disorder before and during lithium treatment (Reference Friedman, Hoau and LevinsonFriedman et al, 1993). The ratios of platelet membrane-bound to cytosolic PKC activities were elevated in the manic group, in whom serotonin-elicited platelet PKC translocation was also enhanced. With respect to brain tissue, Wang & Friedman (Reference Wang and Friedman1996) measured PKC isozyme levels, activity and translocation in post-mortem brain tissue from patients who had bipolar disorder; PKC activity and translocation were greater than in control samples, as were cortical levels of selected PKC isozymes.
In view of the pivotal role of the PKC signalling pathway in the regulation of neuronal excitability, neurotransmitter release and long-term synaptic events (Reference NishizukaNishizuka, 1992; Reference Stabel and ParkerStabel & Parker, 1991; Reference HuangHuang, 1989; Reference Conn, Sweatt and KuoConn & Sweatt, 1994), it was postulated that the attenuation of PKC activity may have a major role in the antimanic effects of lithium and valproate. There was thus a clear need to investigate the efficacy of PKC inhibitors in the treatment of mania. There is currently only one relatively selective PKC inhibitor available for human use — tamoxifen. This is a synthetic, nonsteroidal antioestrogen widely used in the treatment of breast cancer (Reference Catherino and JordanCatherino & Jordan, 1993; Reference JordanJordan, 1994). A number of the effects of tamoxifen are due to oestrogen-receptor antagonism (Reference JordanJordan, 1994), but it has become clear in recent years that it is also a potent PKC inhibitor at therapeutically relevant concentrations (Reference Couldwell, Weiss and DeGiorgioCouldwell et al, 1993). A pilot study investigating the efficacy of tamoxifen in the treatment of acute mania found that this agent does indeed possess antimanic efficacy (Reference Bebchuk, Arfken and Dolan-ManjiBebchuk et al, 2000) (Fig. 3). Despite the small sample size, the results are intriguing, and support the hypothesis that the antimanic effects of lithium and valproate are mediated by PKC inhibition. In view of the apparent involvement of the PKC signalling system in the pathophysiology of bipolar disorder (Reference Friedman, Hoau and LevinsonFriedman et al, 1993; Reference Wang and FriedmanWang & Friedman, 1996), these results indicate that PKC inhibitors may be useful agents in its treatment. Larger, double-blind, placebo-controlled studies of tamoxifen and of novel selective PKC inhibitors in the treatment of mania are clearly warranted.

Fig. 3 Effects of tamoxifen in the treatment of acute mania. Tamoxifen was administered to 10 subjects with acute mania in blinded form; raters were blind to the treatment regimen. Main outcome measures included the Young Mania Rating Scale (YMRS), the Clinician Administered Rating Scale for Mania and the Hamilton Rating Scale for Depression. Tamoxifen resulted in a significant decrease in manic symptomatology rated by the YMRS (* P < 0.05).
Figure reproduced, with permission, from data in Bebchuk et al (Reference Bebchuk, Arfken and Dolan-Manji2000).
EFFECTS OF MOOD STABILISERS ON TRANSCRIPTION FACTORS AND GENE EXPRESSION
It has become increasingly appreciated that biochemical models proposed for the effects of many psychotropic drugs (including mood stabilisers, antidepressants and anti-psychotics) must attempt to account for their special temporal clinical profile — in particular, that the therapeutic effects require a lag period for onset of action, and are generally not immediately reversed upon discontinuation (Reference Hyman and NestlerHyman & Nestler, 1996; Reference Duman, Heninger and NestlerDuman et al, 1997; Reference JopeJope, 1999; Manji et al, Reference Manji, Moore and Chen1999, Reference Manji, Chen, Hsiao, Manji, Bowden and Belmaker2000a ). Patterns of effects requiring such prolonged administration of the drug suggest alterations at the genomic level (Reference Manji, Potter and LenoxManji et al, 1995; Human & Nestler, 1996; Reference Duman, Heninger and NestlerDuman et al, 1997). To investigate the putative effects of mood stabilisers on gene expression, their effects on the DNA binding activity of transcription factors, especially the activator protein 1 (AP-1) family of transcription factors, were examined. The AP-1 factors are a collection of homodimeric and heterodimeric complexes composed of products from two transcription factor families, Fos and Jun. These products bind to a common DNA site, known as the 12-O-tetradecanoyl-phorbol-13-acetate (TPA) response element (TRE), in the regulatory domain of the gene, and activate gene transcription in response to PKC activators, growth factors, cytokines and other agents (including neurotransmitters) (Reference Karin and SmealKarin & Smeal, 1992; Reference Hughes and DragunowHughes & Dragunow, 1995). Induction of c-fos is rapid and transient: Fos protein reaches its maximal levels within 30 minutes, and decreases to low levels within 1-2 hours (Reference Hughes and DragunowHughes & Dragunow, 1995). Induction of c-jun is longer-lasting and varies from a few hours to several days, depending on the cell type and the stimulus (Reference Hughes and DragunowHughes & Dragunow, 1995). The c-jun gene is subject to positive autoregulation through an AP-1 binding site in its promoter. The genes known to be regulated by the AP-1 family of transcription factors in the brain include genes for various neuropeptides, neurotrophins, receptors, transcription factors, enzymes involved in neurotransmitter biosynthesis and proteins that bind to cytoskeletal elements (Reference Hughes and DragunowHughes & Dragunow, 1995).
Several independent laboratories have demonstrated that lithium, at therapeutically relevant concentrations, regulates AP-1 DNA binding activity (Reference Williams and JopeWilliams & Jope, 1995; Reference Jope and SongJope & Song, 1997; Reference Ozaki and ChuangOzaki & Chuang, 1997; Reference Unlap and JopeUnlap & Jope, 1997; Reference Yuan, Chen and ManjiYuan et al, 1998; Reference Chen, Yuan and JiangChen et al, 1999a ). The effects of lithium and valproate on the DNA binding activity of AP-1 were examined using methods verified by both competition assay with cold and mutant TRE oligos and supershift assay with antibodies against AP-1 transcription factors (Reference Yuan, Chen and ManjiYuan et al, 1998; Reference Chen, Yuan and JiangChen et al, 1999a ). Both lithium and valproate at therapeutically relevant concentrations, produced a time- and concentration-dependent increase in the DNA binding of TRE to AP-1 transcription in rat brain ex vivo, and in cultured human neuroblastoma cells factors (Reference Yuan, Chen and ManjiYuan et al, 1998; Reference Chen, Yuan and JiangChen et al, 1999a ). It was further confirmed that these effects on AP-1 DNA binding activity do, in fact, translate into changes at the gene expression level. Effects of lithium on gene expression were first studied in cells transiently transfected with the pGL2 control vector. The reporter gene, luciferase in the pGL2 control vector, is driven by simian virus 40 (SV40) promoter, which has two characterised AP-1 sites. Using this reporter gene transfection system, the role of the AP-1 sites in mediating valproate's effects on gene expression was further examined by eliminating the AP-1 sites by mutagenesis. Lithium increased the expression of the luciferase reporter gene driven by an SV40 promoter/enhancer containing TREs (Reference Yuan, Chen and ManjiYuan et al, 1998; Reference Chen, Yuan and JiangChen et al, 1999a ); this increase was time- and concentration-dependent. Furthermore, mutations in the TRE sites of the reporter gene promoter markedly attenuated these effects. These data indicate that lithium may stimulate gene expression (at least in part) through the AP-1 transcription factor pathway, effects that may play an important part in its long-term clinical actions (Fig. 4).

Fig. 4 Effects of lithium and valproate (VPA) on luciferase reporter gene activity in cultured cells: (a) time and (b) dose dependency. Cells were cultured, transfected with the pGL2 control vector and then incubated with lithium or valproate at the concentrations indicated in (a) for 24 hours, or with lithium (1.0 mol/l) or VPA (1.0 mol/l) for the times indicated in (b) as described in Yuan et al (Reference Yuan, Chen and Manji1998) and Chen et al (Reference Chen, Yuan and Jiang1999a ). Luciferase activity was assayed in the whole cell lysates using the Promega kit. Data are means (s.e.). * P < 0.05 compared to control. (c) Attenuation of the lithium- or valproate-induced increases in luciferase activity by TRE mutations. The mutations on the TRE sites were made using the QuikChange site directed mutagenesis kit. Cells were transfected with either pGL2 control or mutant pGL2 control and then incubated with 1.0 mmol/l lithium or 0.6 mmol/l valproate for 24 hours. Luciferase activity was assayed using the Promega kit. The values in the bar graphs are the means (s.e.) of three or more experiments. Reproduced from Yuan et al (Reference Yuan, Chen and Manji1998) and Chen et al (Reference Chen, Zeng and Jiang1999c ) with permission.
To ascribe therapeutic relevance to the observed biochemical findings it is necessary to demonstrate that they do, in fact, also occur in critical regions of the central nervous system in vivo. It is well established that the expression of tyrosine hydroxylase (TH) is largely mediated by the AP-1 family of transcription factors (Reference Kumer and VranaKumer & Vrana, 1996). The effects of acute and chronic lithium on the levels of TH in three brain areas that have been implicated in the pathophysiology of mood disorders — the frontal cortex, hippocampus and striatum — were therefore examined (Reference Goodwin and JamisonGoodwin & Jamison, 1990; Drevets et al, Reference Drevets, Videen and Price1992, Reference Drevets, Price and Simpson1997; Reference Ketter, George, Kimbrell, Joffe and YoungKetter et al, 1997; Reference Soares and MannSoares & Mann, 1997). It was found that chronic lithium treatment significantly increased the levels of TH in all three brain areas (Reference Chen, Yuan and JiangChen et al, 1998). Future in situ hybridisation studies or immunohistochemistry studies are needed to determine if lithium also increases the expression of TH in areas of brain known to contain the cell bodies of the major noradrenergic and dopaminergic systems, namely the locus caeruleus, ventral tegmental area and substantia nigra. The results clearly show that in addition to increasing AP-1 DNA binding activity and the expression of the luciferase reporter gene in vitro, chronic lithium administration increases the TH levels in areas of rat brain ex vivo. These effects are compatible with an effect on the DNA binding of TRE to the AP-1 family of transcription factors, and have the potential to regulate patterns of gene expression in critical neuronal circuits (Reference Boyle, Smeal and DefizeBoyle et al, 1991; Reference Lin, Smeal and BineturyLin et al, 1993). In view of the key roles of these nuclear transcription regulatory factors in longterm neuronal plasticity and cellular responsiveness, and the potential to regulate patterns of gene expression in critical neuronal circuits (Reference Boyle, Smeal and DefizeBoyle et al, 1991; Reference Lin, Smeal and BineturyLin et al, 1993), these effects may be important in the therapeutic efficacy of lithium and valproate, and are worthy of further study. The precise mechanisms by which lithium regulates AP-1 DNA binding activity still need to be fully delineated, but may involve effects on PKC isozymes (Reference Yuan, Chen and ManjiYuan et al, 1999). Furthermore, as we discuss in greater detail below, lithium, at clinically relevant concentrations, has been demonstrated to inhibit the activity of glycogen synthase kinase-3β (GSK-3β) (Hedgepeth et al, 1997; Reference Klein and MeltonKlein & Melton, 1996; Reference Stambolic, Ruel and WoodgettStambolic et al, 1996). This enzyme is known to phosphorylate c-jun at three sites adjacent to the DNA binding domain, thereby reducing TRE binding (Reference Lin, Smeal and BineturyLin et al, 1993; Reference Stambolic, Ruel and WoodgettStambolic et al, 1996).
GLYCOGEN SYNTHASE KINASE
Is GSK-3β a therapeutically relevant target for mood-stabilising agents?
In the late 1990s a completely unexpected target for the action of lithium was identified. Klein & Melton (Reference Klein and Melton1996) were the first to demonstrate that lithium, at therapeutically relevant concentrations, is an inhibitor of GSK-3β. This is an evolutionarily highly conserved kinase, originally identified as a regulator of glycogen synthesis, and is now known to have an important role in the central nervous system, by regulating various cytoskeletal processes through its effects on tau and synapsin I, as well as long-term nuclear events by phosphorylation of c-jun, and nuclear translocation of β-catenin (Reference Klein and MeltonKlein & Melton, 1996; Reference Wagner, Utton and GalloWagner et al, 1996; Reference Yost, Torres and MillerYost et al, 1996; Reference Ikeda, Kishida and YamamotoIkeda et al, 1997; Reference Lucas and SalinasLucas & Salinas, 1997). However, lithium is known to bring about a variety of biochemical effects (Reference Bunney, Garland-Bunney and MeltzerBunney & Garland-Bunney, 1987; Reference Mork, Geisler and HollundMork et al, 1992; Reference Chang, Grange and RabinChang et al, 1996; Reference JopeJope, 1999) (see Table 2), and it is unclear if inhibition of GSK-3β is therapeutically relevant. A series of studies were therefore undertaken to determine if valproate, like lithium, also regulates GSK-3; it was found that valproate inhibits both GSK-3α and GSK-3β in a concentration-dependent manner, with significant effects observed at concentrations similar to those attained clinically (Reference Chen, Huang and JiangChen et al, 1999b ) (Fig. 5).

Fig. 5 Effects of valproate (VPA) on glycogen synthase kinase 3 (GSK-3). (a, b) Effects of VPA on GSK-3α activity. The reaction was carried out using purified or recombinant GSK-3, and 150 μmol/l MgATP (as described in Reference Chen, Huang and JiangChen et al, 1999b ) with VPA, either in the absence (a) or presence (b) of additional Mg2+ (20 mmol/l). Data are means (s.e.) from three experiments. (c, d) Effects of VPA on GSK-3β activity (as described in Reference Chen, Huang and JiangChen et al, 1999b ) in the absence (c) or presence (d) of additional Mg2+ (20 mmol/l). Data are means (s.e.) from three experiments. VPA inhibited GSK-3α and GSK-3β in a concentration-dependent manner, both in the absence and presence of Mg2+; * P < 0.05 compared to control (e, f). Additivity of the effects of VPA and lithium on GSK-3 activity. The reaction was carried out at room temperature for 20 minutes utilising 0.5 U GSK-3α or GSK-3β, with either VPA alone (0.6 mmol/l), or VPA (0.6 mmol/l) plus lithium (1.0 mmol/l). Data are means (s.e.) from three experiments. The addition of lithium resulted in further significant reductions in the activity of both GSK-3α and GSK-3β; * P < 0.05 compared to VPA alone. Figure modified, and reproduced, with permission from Chen et al (Reference Chen, Huang and Jiang1999b ).
Table 2 Biochemical mediators of lithium's effects in the central nervous system: a surfeit of candidates

| Na+/K+ ATPase |
| Neurotransmitters |
| Serotonin |
| Dopamine |
| Noradrenalin |
| Acetylcholine |
| Glutamate |
| Receptors |
| β, α2, D2, 5-HT1A |
| Signalling |
| Phosphoinositides |
| PKC, MARCKS |
| G proteins |
| Phospholipase A2 |
| Adenylate cyclases |
| Calcium |
| GSK-3β |
| Gene expression |
Incubation of intact human neuroblastoma SH-SY5Y cells with valproate resulted in an increase in the subsequent in vitro recombinant GSK-3β-mediated 32P incorporation into two putative GSK-3 substrates (approximate molecular weights 85 and 200), compatible with inhibition of endogenous GSK-3β by valproate. Consistent with GSK-3β inhibition, incubation of SH-SY5Y cells with valproate results in a significant time-dependent increase in both cytosolic and nuclear β -catenin levels. Since GSK-3β plays a critical role in the central nervous system by regulating various cytoskeletal processes as well as long-term nuclear events, and is a common target for both lithium and valproate, its inhibition may underlie some of the long-term therapeutic effects of mood-stabilising agents, and represents an exciting area of future research.
MESSENGER RNA DIFFERENTIAL DISPLAY STUDIES
Regulation of long-term changes in gene expression by mood-stabilising agents
Although several specific genes that are the targets of long-term lithium and/or valproate have been identified, it has been estimated that approximately 10 000-15 000 genes may be expressed in a given cell at any time, and thus new methods are required to study the complex pattern of gene expression changes induced by chronic drug treatment (Reference Hyman and NestlerHyman & Nestler, 1996; Reference JopeJope, 1999; Manji et al, Reference Manji, Moore and Chen1999, Reference Manji, Moore and Rajkowska2000b ). New methodologies have evolved to identify the differential expression of multiple genes (e.g. in pathological v. normal tissue, or in control v. treated tissue). The pool of transcribed mRNAs in a given cell is termed a ‘transcriptome’, and underlies the cell phenotype. The transcriptome, which includes the transcribed genes and the corresponding number of mRNA copies, changes under both physiological and pathological conditions, as well as in response to treatments. Identifying the genes whose transcription is altered in the transition from one functional state to another is central to defining the precise molecular events that regulate cell phenotype and function. A series of studies utilising the technique of mRNA differential display, based on reverse transcription and the polymerase chain reaction, to identify changes in gene expression associated with the therapeutic efficacy of mood stabilisers, investigated the effects of lithium and valproate (the only two medications approved by the US Food and Drug Administration for the treatment of bipolar disorder). Although these two agents are unlikely to exert their therapeutic effects by precisely the same mechanisms, identifying the genes that are regulated in concert by these structurally highly dissimilar agents may provide important clues to the molecular mechanisms underlying mood stabilisation (Reference Ikonomov and ManjiIkonomov & Manji, 1999). Inbred male Wistar Kyoto rats (selected to reduce potential false positives due to individual differences) were treated chronically with lithium, valproate or saline. Ribonucleic acid was extracted from the frontal cortices to study gene expression using mRNA differential display (Reference Liang and PardeeLiang & Pardee, 1992; Reference Liang, Bauer and AverboukhLiang et al, 1995). One hundred and fifty reactions were performed with the combination of three one-base anchored 3′ primers and fifty 5′ arbitrary primers (Reference Liang and PardeeLiang & Pardee, 1992; Reference Liang, Bauer and AverboukhLiang et al, 1995); on average, 270 bands were obtained in each reaction. Clones were sequenced from T7 and M13 priming sites using an automated sequencer; BLAST searches were conducted for each clone obtained, and the homology with known sequences in the GenBank database was further evaluated using the BESTFIT program from the Genetic Computer Group's sequence analysis software package. To date, we have identified a number of mRNA species whose expression is markedly increased or decreased by both treatments.
One of the genes whose expression is markedly increased by both lithium and valproate (clone 12; GenBank accession number AF087437) is 355 base pairs long, contains a poly(A)tail, and shows high sequence homology with the 3′ end of the β subunit of the mouse (92% identical sequences) and human (85%) transcription factor, polyomavirus enhancer binding protein 2 (PEBP2)β, also known as core-binding factor (CBF) β and acute myelogenous leukaemia 1 (AML1) β (Reference Liu, Tarle and HajraLiu et al, 1993; Reference Wang, Stacy and MillerWang et al, 1996; Reference Kanno, Kanno and ChemKanno et al, 1998). In the absence of available antibodies to PEBP2-β, we next sought to determine if the treatments induced functional changes in PEBP2 transcription factor activity. Treatment with both lithium and valproate increased the DNA binding activity of PEBP2-αβ in the frontal cortex (Reference Chen, Zeng and JiangChen et al, 1999c ). To determine if these effects are specific for mood stabilisers, we investigated the effects of chronic D-amphetamine sulphate and chlordiazepoxide; neither of these treatments produced any detectable changes in PEBP2-αβ DNA binding activity. We therefore investigated putative targets of the PEBP2 transcription factor, which may be of therapeutic relevance in the treatment of bipolar disorder.
The promoter of the B-cell lymphoma protein 2 (bcl-2) gene (both rat and human genes) has a PEBP2 binding site, and this site has been clearly demonstrated to increase the expression of a reporter gene driven by the bcl-2 promoter (Reference Klampfer, Zhang and ZelenetzKlampfer et al, 1996). A neuroprotective role for bcl-2 is well established, and constitutive expression of high levels of bcl-2 protein enhances the survival of cells when exposed to adverse stimuli (Reference Jacobson and RaffJacobson & Raff, 1995; Reference Merry and KorsmeyerMerry & Korsmeyer, 1997). Additionally, the delivery in vivo of a bcl-2 expression vector protects neurons against focal ischaemia (Reference Lawrence, Ho and SunLawrence et al, 1996), and bcl-2 has also been demonstrated to promote neuronal regeneration (Reference Chen, Yuan and HawverChen et al, 1997). In this context, it is noteworthy that mood disorders, including bipolar disorder, are associated with volumetric changes on magnetic resonance imaging and computed tomography, suggestive of neuronal atrophy or loss (Reference Elkis, Friedman and MeltzerElkis et al, 1995; Reference Drevets, Price and SimpsonDrevets et al, 1997; Reference Ketter, George, Kimbrell, Joffe and YoungKetter et al, 1997). Furthermore, both brain imaging studies and postmortem morphometric three-dimensional cell counting studies have implicated the frontal cortex as a site of neuronal atrophy or loss in bipolar disorder (Dreets et al, 1997; Reference Rajkowska, Miguel-Hidalgo and WeiRajkowska et al, 1999; Reference RajkowskaRajkowska, 2000). We therefore investigated whether the treatment-induced increase in PEBP2-αβ DNA binding activity in the frontal cortex was accompanied by changes in the levels of the neuroprotective protein, bcl-2. Chronic treatment of rats with both lithium and valproate resulted in a doubling of bcl-2 levels in the frontal cortex, effects that were accompanied by a marked increase in the number of bcl-2 immunoreactive cells in layers 2 and 3 of the frontal cortex. It is noteworthy that neuroprotective effects have recently been reported for both lithium and valproate (Reference Bruno, Sortino and ScapagniniBruno et al, 1995; Reference Nonaka, Hough and ChuangNonaka et al, 1998); the robust increases in the levels of bcl-2 that we have demonstrated may have a major role in mediating these effects, and suggest that mood-stabilising agents may bring about some of their long-term beneficial effects via hitherto underappreciated neuroprotective effects (Reference Smith, Makino and AltemusSmith et al, 1995; Reference Duman, Heninger and NestlerDuman et al, 1997; Reference Manji, Moore and ChenManji et al, 1999).
CONCLUSION
Regulation of signal transduction and gene expression within critical regions of the brain affects the intracellular signal generated by a number of neurotransmitter systems; these effects thus represent attractive putative mediators of the therapeutic actions of mood stabilisers, effects that are probably mediated via their effects on a network of interconnected neurotransmitter pathways (Fig. 6). It is becoming increasingly clear that for many refractory patients with these disorders, new drugs simply mimicking the ‘traditional’ drugs that directly or indirectly alter neurotransmitter levels or bind to cell surface receptors may be of limited benefit. This is because such strategies implicitly assume that the target receptors are functionally intact, and that altered synaptic activity will thus be transduced to modify the postsynaptic ‘throughput’ of the system. However, the possible existence of abnormalities in signal transduction pathways suggests that for patients refractory to conventional medications, benefit may only be obtained by the direct targeting of post-receptor sites (Reference NestlerNestler, 1998; Manji et al, Reference Manji, Chen, Hsiao, Manji, Bowden and Belmaker2000a , Reference Manji, Drevets and Charney2001). Recent discoveries concerning a variety of mechanisms involved in the formation and inactivation of second messengers offer the promise for the development of novel pharmacological agents designed to ‘site-specifically’ target signal transduction pathways. Although clearly more complex than the development of receptor-specific drugs, it may be possible to design novel agents to selectively affect second-messenger systems because they are heterogeneous at the molecular and cellular level, are linked to receptors in a variety of ways, and are expressed in different stoichiometries in different cell types. Additionally, since signal transduction pathways display certain unique characteristics depending on their activity state, e.g. rate of guanine nucleotide exchange, G protein conformational states, GTP hydrolysis, interaction with different regulators of G protein signalling (RGS) proteins, cytosol to membrane translocation of PKC isozymes and receptor kinases, they offer built-in targets for relative specificity of action, depending on the ‘set point’ of the substrate. It is our strong conviction that it is at the cellular and molecular level that some of the most exciting advances in our understanding of the long-term therapeutic action of lithium will take place in the coming years (Manji & Lenox, Reference Manji and Lenox1999, Reference Manji and Lenox2000). Current studies of long-term lithium-induced changes to the PKC signalling pathway (including PKC isozyme regulation, post-translational modification of important phosphoproteins, and PKC-mediated gene expression) are a most promising avenue for future investigation. An exciting recent finding is the hitherto completely unexpected demonstration that lithium robustly increases the levels of the major neuroprotective protein bcl-2 (Reference Manji, Moore and ChenManji et al, 1999). This upregulation of bcl-2 is accompanied by neurotrophic effects of lithium in both preclinical and clinical studies (Reference Manji, Moore and RajkowskaManji et al, 2000b ; Reference Moore, Bebchuk and HasanatMoore et al, 2000a ,Reference Moore, Bebchuk and Wilds b ). In view of the recent brain imaging studies and postmortem morphometric three-dimensional cell counting studies demonstrating neuronal atrophy or loss in bipolar disorder (Reference Rajkowska, Miguel-Hidalgo and WeiRajkowska et al, 1999), these results raise the intriguing possibility that mood-stabilising agents may bring about some of their long-term beneficial effects by their underappreciated neuroprotective action (Duman et al, Reference Duman, Heninger and Nestler1997, Reference Duman, Malberg and Nakagawa2000; Manji et al, Reference Manji, Moore and Chen1999, Reference Manji, Moore and Rajkowska2000b ) (Fig. 7). These recent observations also suggest that impairments of neuroplasticity and cellular resilience may contribute to the pathophysiology of mood disorders, and the poor long-term outcome observed in many patients (Reference Manji, Moore and RajkowskaManji et al, 2000b ). Strategies are being investigated to develop small molecule switches for protein—protein interactions, which have the potential to regulate the activity of growth factors, MAP kinase cascades and interactions between homodimers and heterodimers of the bcl-2 family of proteins (Reference Guo, Zhou and SchultzGuo et al, 2000); the development of treatments that directly target molecules involved in critical neuronal survival and cell death pathways would have the potential to enhance neuroplasticity and cellular resilience, and thereby modulate the long-term course and trajector of these devastating illnesses.
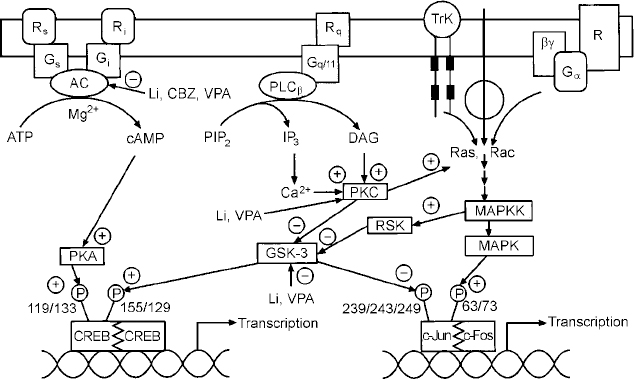
Fig. 6 Effects of mood stabilisers on signalling pathways and gene expression. Abbreviations as follows: receptors coupled to stimulation (Rr) or inhibition (Ri) of adenylate cyclase (AC); G proteins mediating stimulation (G5) or inhibition (Gi) of adenylate cyclase; receptors (Rq) coupled to the G protein, Gqll; phospholipase C β isozyme (PLCβ); receptor tyrosine kinase (TrK); phosphatidylinositol 4,5-bisphosphate (PIP2); diacylglycerol, DAG; inositol 1,4,5-trisphosphate (IP3); protein kinase C (PKC); mitogen activated protein kinase (MAPK); mitogen activated protein kinase kinase (MAPKK); glycogen synthase kinase 3 (GSK-3); cyclic AMP response element binding protein (CREB); lithium (Li); carbamazepine (CBZ); valproate (VPA).
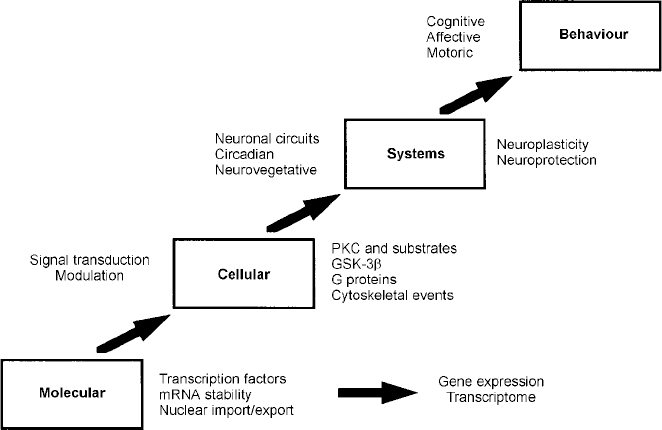
Fig. 7 Four principal levels of lithium effects. Lithium exerts its primary biochemical effects at the molecular and cellular levels. These effects bring about changes in critical interacting neuronal circuits, thereby regulating affective, cognitive and motor systems, effects that are ultimately responsible for bringing about long-term stabilisation of mood. GSK-3β, glycogen synthase kinase 3β; mRNA, messenger RNA; PKC, protein kinase C.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Greater understanding of the molecular and cellular mechanisms of action of mood-stabilising agents will enhance the rationale for the development of new treatments targeting intracellular targets.
-
▪ The long-term therapeutic effects of mood-stabilising agents may involve underappreciated neurotrophic effects.
LIMITATIONS
-
▪ As yet, there are no phenotypical changes in rodent studies that are clearly associated with treatment response.
-
▪ Most of the studies have been conducted in ‘normal’ rodents.
-
▪ Bipolar disorder undoubtedly represents a heterogeneous group of disorders, and current studies do not adequately address this.
ACKNOWLEDGEMENTS
The authors' research is supported in part by the National Institute of Mental Health, the Theodore and Vada Stanley Foundation, National Association for Research on Schizophrenia and Depression and the Joseph Young Senior Foundation. The authors would also like to acknowledge the valuable contributions of Joseph M. Bebchuk, MD; Debra Glitz, MD; the nursing staff of the Neuropsychiatric Research Unit; and Ms Celia Knobelsdorf for providing outstanding editorial assistance.










eLetters
No eLetters have been published for this article.