



Noonan syndrome is a predominantly autosomal dominant disorder with physical, cognitive, and psychosocial features. The estimated incidence rates vary from 1:1000 to 1:2500 live births. Reference Romano, Allanson and Dahlgren1,Reference Roberts, Allanson, Tartaglia and Gelb2 Noonan syndrome is characterised by short stature, typical facial features, thorax deformities, cardiac abnormalities, renal abnormalities, cryptorchidism, delayed motor development, and bleeding disorders. Reference Roberts, Allanson, Tartaglia and Gelb2,Reference Turner3 There is a major variation in the severity and nature of these characteristics. Reference Roberts, Allanson, Tartaglia and Gelb2
Mutations identified in NS are gain-of-function mutations, which result in increased RAS-mitogen-activated protein kinase signalling. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Kouz, Lissewski and Spranger5 The first gene discovered to be responsible for Noonan syndrome was PTPN11 on chromosome 12q. Reference Tartaglia, Mehler and Goldberg6 In recent years, more genes that cause Noonan syndrome has been discovered. Mutations in PTPN11 (40–67%), SOS1 (10–26%), and RAF1 (3–17%) are most common. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Kouz, Lissewski and Spranger5,Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Allanson, Bohring and Dorr8 Other genes involved in Noonan syndrome are KRAS (1–5%), NRAS, RIT1 (3–7%), RRAS, CBL, SOS2, LZTR1, and MAP2K1. Reference Kouz, Lissewski and Spranger5,Reference Allanson, Bohring and Dorr8–Reference Allanson, Roberts, Adam, Ardinger, Pagon, Wallace, Bean and Stephens10 In 18–24% of the patients with Noonan syndrome, no gene mutation is found. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Allanson, Roberts, Adam, Ardinger, Pagon, Wallace, Bean and Stephens10 For this reason, a clinical scoring system has been developed with major and minor features. Reference van der Burgt11,Reference van der Burgt, Berends, Lommen, van Beersum, Hamel and Mariman12 Major cardiac features in this scoring system are pulmonary stenosis, hypertrophic cardiomyopathy, and/or an electrocardiogram typical for NS. Minor cardiac features are “other cardiac defects”.
The electrocardiogram in patients with NS often shows typical features (Fig 1). These typical Noonan syndrome-related electrocardiographic features are left axis deviation, small R-waves in the left precordial leads, large S-waves in the right precordial leads, abnormal Q-waves, and/or wide QRS complex. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13–Reference Bertola, Kim and Sugayama15 These typical electrocardiographic abnormalities are present in 50–60% of all patients with Noonan syndrome, and do not seem to be associated with the presence of a (specific) CHD and the presence of a PTPN11 mutation. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13 The reason for these characteristic electrocardiographic features is still unknown. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13,Reference Bertola, Kim and Sugayama15
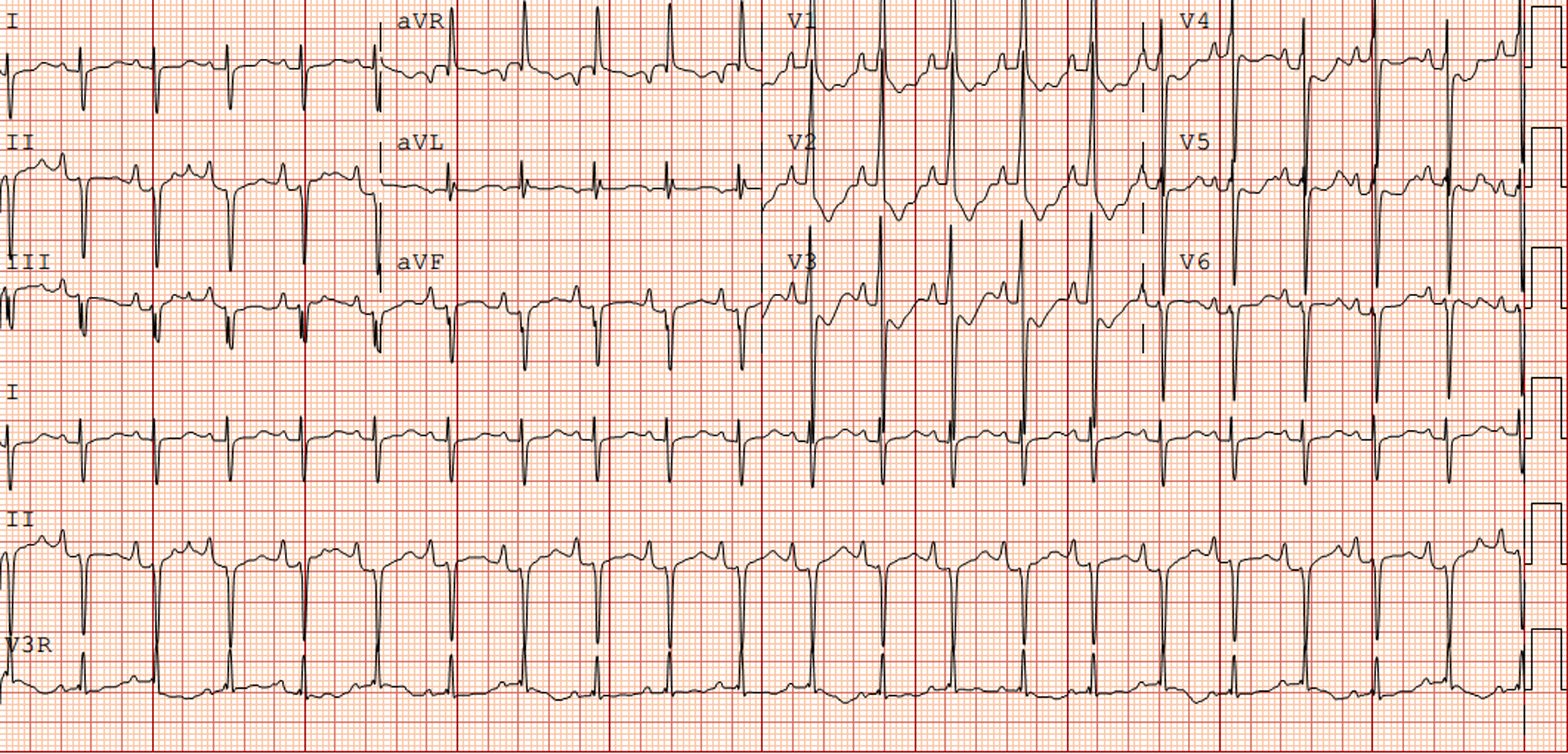
Figure 1. Electrocardiogram (speed 25 mm/sec, voltage 0.1 mV/mm) from a newborn with Noonan syndrome due to PTPN11 mutation with a severe pulmonary valve stenosis.
More than 60% of NS patients have CHDs. The CHD most commonly seen are pulmonary valve stenosis and hypertrophic cardiomyopathy. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Jongmans, Sistermans and Rikken16 Other CHD seen in Noonan syndrome are atrial septal defects, ventricular septal defects, patent ductus arteriosus, aortic coarctation, and mitral valve abnormalities. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Yaoita, Niihori and Mizuno9,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13 Recent studies show that PS is more often associated with mutations in PTPN11 (45–65%), SOS1 (50–83%), BRAF (75%), and RIT1 (77%), while HCM is associated with mutations in RAF1 (75 –100%) and RIT1 (42–56%). Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Kouz, Lissewski and Spranger5,Reference Yaoita, Niihori and Mizuno9,Reference Ko, Kim, Kim and Yoo17
The purpose of this study is to analyse whether the typical Noonan syndrome-related electrocardiographic characteristics can still be seen as a major feature even with modern echocardiographic possibilities and genetic tests, for example, in patients with clinical Noonan syndrome with a gene variant of unknown significance.
In addition, we investigated whether the typical electrocardiographic features in patients with Noonan syndrome are dependent on the genotype or type of CHD in these patients.
Materials and methods
Patients
In this single-centre retrospective study, we analysed all patients seen in the outpatient department of the Noonan Syndrome Expertise Centre of the Radboud University Medical Center before 2019 with clinically and genetically confirmed Noonan syndrome. The clinical diagnosis of Noonan syndrome was based on the earlier mentioned scoring system of van der Burgt et al and confirmed by DNA analysis. Reference van der Burgt11,Reference van der Burgt, Berends, Lommen, van Beersum, Hamel and Mariman12 Patients without an available electrocardiogram or echocardiogram before the surgical intervention or balloon valvuloplasty were excluded from the study. According to the Medical Ethics Committee of the district Arnhem/Nijmegen, no ethical approval was required for this study (file number 2020-6851).
Electrocardiogram
Specific electrocardiographic features included in this study were left axis deviation, large S-waves in the right precordial leads, small R-waves in the left precordial leads, abnormal Q-waves, and/or wide QRS complexes. The features were reviewed on the first ECG available using the following definitions:
-
Left axis deviation: QRS axis is less than the lower limit of normal for the patient’s age. Reference Davignon, Rautaharju, Boisselle, Soumis, Mégélas and Choquette18
-
Large S-waves in V2: S-wave is more than the upper limit of normal for the patient’s age. Reference Park19,Reference Park and Guntheroth20
-
Small R-waves in V5V6: little R deflection over the left precordium with an R/S ratio is lower than the lowest limits of normal and R voltage in V5 and V6 is less than 50% of the mean according to Park and Guntheroth. Reference Park and Guntheroth20
-
Abnormal Q-wave: Q voltage is greater than the upper limit of normal and wider than 0.04 seconds.
-
Wide QRS complex: QRS duration of more than 0.08 seconds under the age of 3 years, above 0.10 seconds between 3 and 12 years, and above the upper limit for QRS duration of 0.12 seconds from 12 years and older.
Echocardiogram
Diagnosis of CHD defect included in this study was established by the first echocardiogram available and this had to be performed before balloon valvuloplasty or surgical intervention. In general, the guidelines and standards for the performance of paediatric echocardiograms were used. Reference Lai, Geva and Shirali21 Whenever possible, the original recordings were used for diagnosis; otherwise, the recorded diagnosis was used. Pulmonary stenosis was classified as mild (peak gradient <36 mmHg), moderate (peak gradient 36–64 mmHg), and severe (peak gradient >64 mmHg). Hypertrophic cardiomyopathy was diagnosed according to the criteria used by Wilkinson et al. Reference Lai, Geva and Shirali21
Statistical analysis
All analyses were carried out using SPSS, version 22.0 (IBM Corp., NY, United States of America). Results are expressed as median with interquartile range, and percentages. Statistical significance was analysed using Fisher’s exact test as appropriate with a significance threshold of p ≤ 0.05.
Results
Demographics
A cohort of 95 patients was included in this study. Of these patients, 46 (48.4%) were male and 49 (51.6%) were female, and their median (IQR) age at the date of the ECG was 4.9 years (0.9–14.4). Table 1 shows the distribution of the involved genes, the cardiac defects, and typical Noonan syndrome-related electrocardiographic features. Sixty (63.2%) of the patients had a mutation in PTPN11, and mutations in SOS1 and KRAS were found in 8 (8.4%) patients.
Table 1. Cardiac heart defects and ECG abnormalities in patients with Noonan syndrome
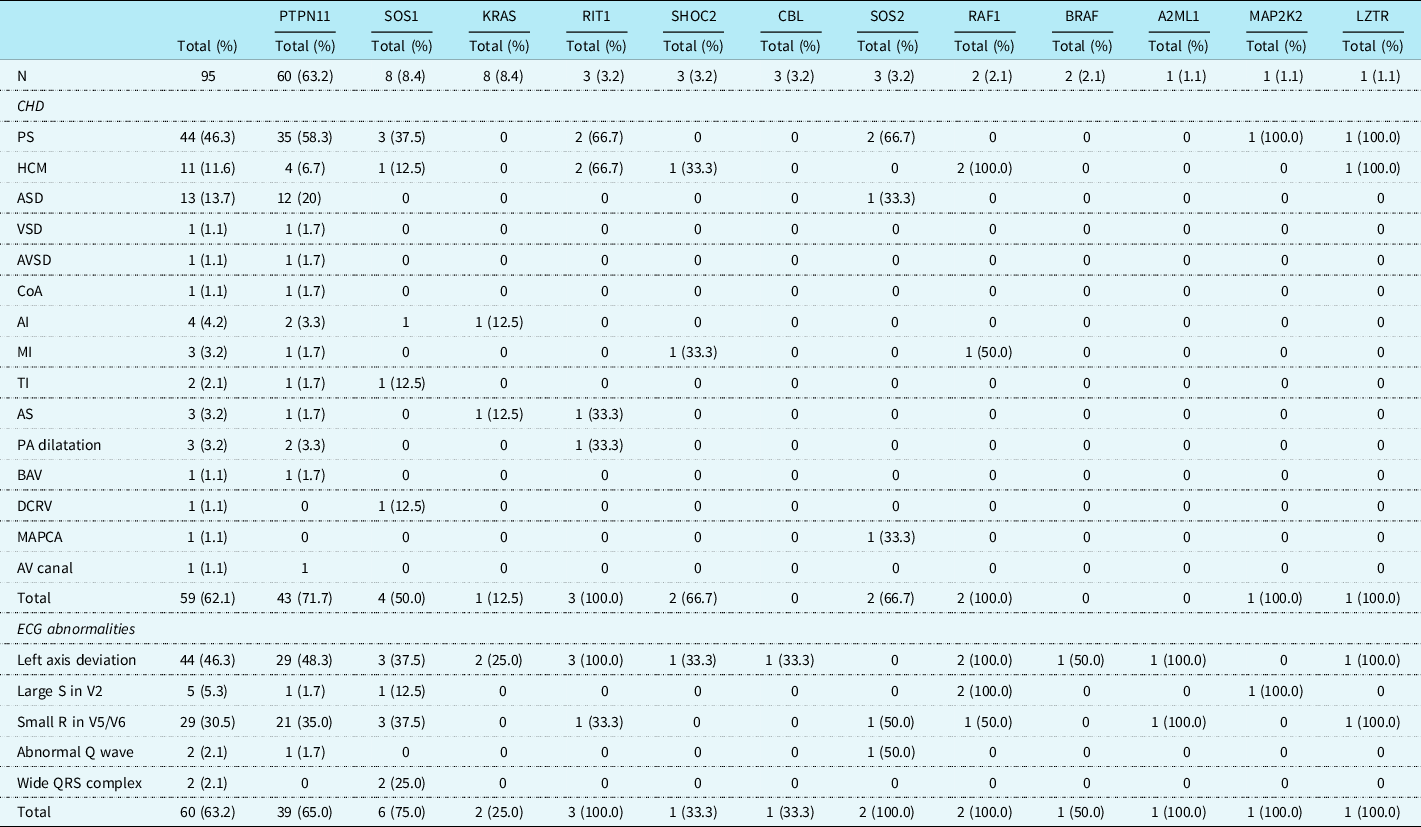
AI = aortic insufficiency; AS = aortic stenosis; ASD = atrial septal defect; AV = atrioventricular; AVSD = atrial ventricular septal defect; BAV = bicuspid aortic valve; CHD = congenital heart defect; CoA = coarctatio aortae; DCRV = double-chambered right ventricle; ECG = electrocardiogram; HCM = hypertrophic cardiomyopathy; MAPCA = major aortopulmonary collateral artery; MI = mitral insufficiency; PA = pulmonary artery; PS = pulmonary stenosis; TI = tricuspid insufficiency; VSD = ventricular septal defect.
Congenital heart defects
It was possible to use the initial echocardiographic recordings in 55 of the 95 patients. In 40 patients, the recorded diagnosis was used. Fifty-nine (62.1%) patients had CHD. Pulmonary valve stenosis (mild in 53%, moderate 11%, and severe in 36% of the cases; all severe cases had a therapeutic intervention afterwards) was most frequently seen followed by an atrial septal defect (haemodynamically important in 35%, not important 65%) and hypertrophic cardiomyopathy (of which two needed a surgical intervention afterwards), in, respectively, 44 (46.3%), 13 (13.7%), and 11 (11.6%) patients. Pulmonary valve stenosis was most often seen in patients with a mutation in PTPN11 (58.3%), SOS1 (37.5%), RIT1 (66.7%), and SOS2 (66.7%). Patients with a PTPN11 gene mutation had a significantly higher incidence of pulmonary valve stenosis (58.3%) and atrial septal defect (20%) than patients with other gene mutations (respectively, p = 0.003 and p = 0.027). Hypertrophic cardiomyopathy was more frequently diagnosed in patients with mutations of RIT1 (66.7%) and RAF1 (66.7%) genes, which was significantly more frequently than in the other NS genes (respectively, p = 0.035 and p = 0.012).
Electrographic abnormalities
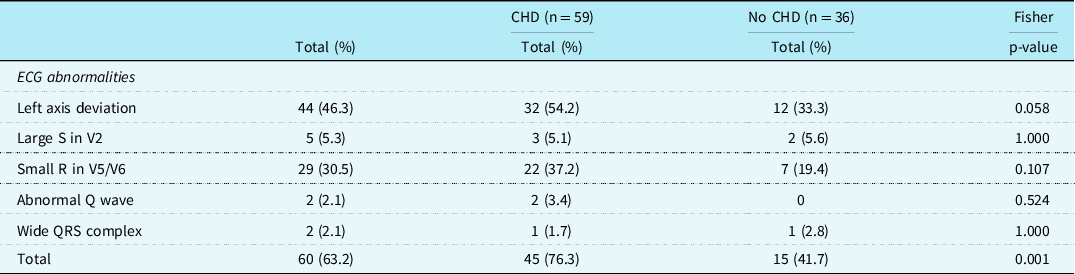
In 60 patients (63.2%), one or more typical Noonan syndrome-related electrocardiographic features were seen. Most of the patients with CHD did show these typical Noonan syndrome-related electrocardiographic features (45/59, 76.3%). Also, in 15 of the 36 (41.7%) patients with a structurally normal heart on echocardiogram, typical Noonan syndrome electrocardiographic abnormalities were detected (Table 1). There was no significant difference in the incidence of the individual-specific Noonan syndrome-related electrocardiographic features between the group with CHD and the group without CHD, however, specific electrocardiographic features as a group occurred significantly more frequently in patients with CHD (p = 0.001).
There were 44 (46.3%) patients with left axis deviation, 29 (30.5%) patients with small R-waves in the left precordial leads, and 5 (5.3%) patients with large right precordial S-waves. An abnormal Q-wave was seen in 2 (2.1%) patients and a wide QRS complex also in 2 (2.1%) patients (Table 2). Typical Noonan syndrome-related electrographic features were not more often seen in patients with a mutation in the PTPN11 gene than in patients with other gene mutations. Patients with a mutation in the RAF1 gene had more frequently large S-waves in the right precordial leads (p = 0.002), and patients with SOS1 gene mutation had more frequent wide QRS complexes (p = 0.006).
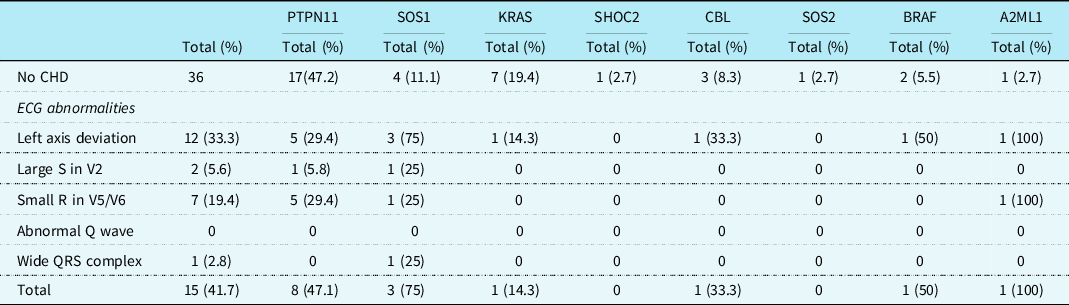
Table 2. Specific ECG features in patients with Noonan syndrome with and without CHD
Regarding the 36 patients without a CHD, 15 (41.7%) had specific Noonan syndrome-related electrocardiographic features. These electrocardiographic features were seen in patients with mutations in PTPN11, SOS1, KRAS, CBL, BRAF, and A2ML1 genes (Table 3). Left axis deviation was seen in 12 (33.3%) patients, small left precordial R-waves in 7 (19.4%) patients, large right precordial S-waves leads in 2 (5.6%) patients, and wide QRS complex in 1 (2.8%) patient (Table 3). One child with a SOS1 mutation without CHD had a complete left bundle branch block with left axis deviation with a septal flash on echocardiography (Fig 2a and b).
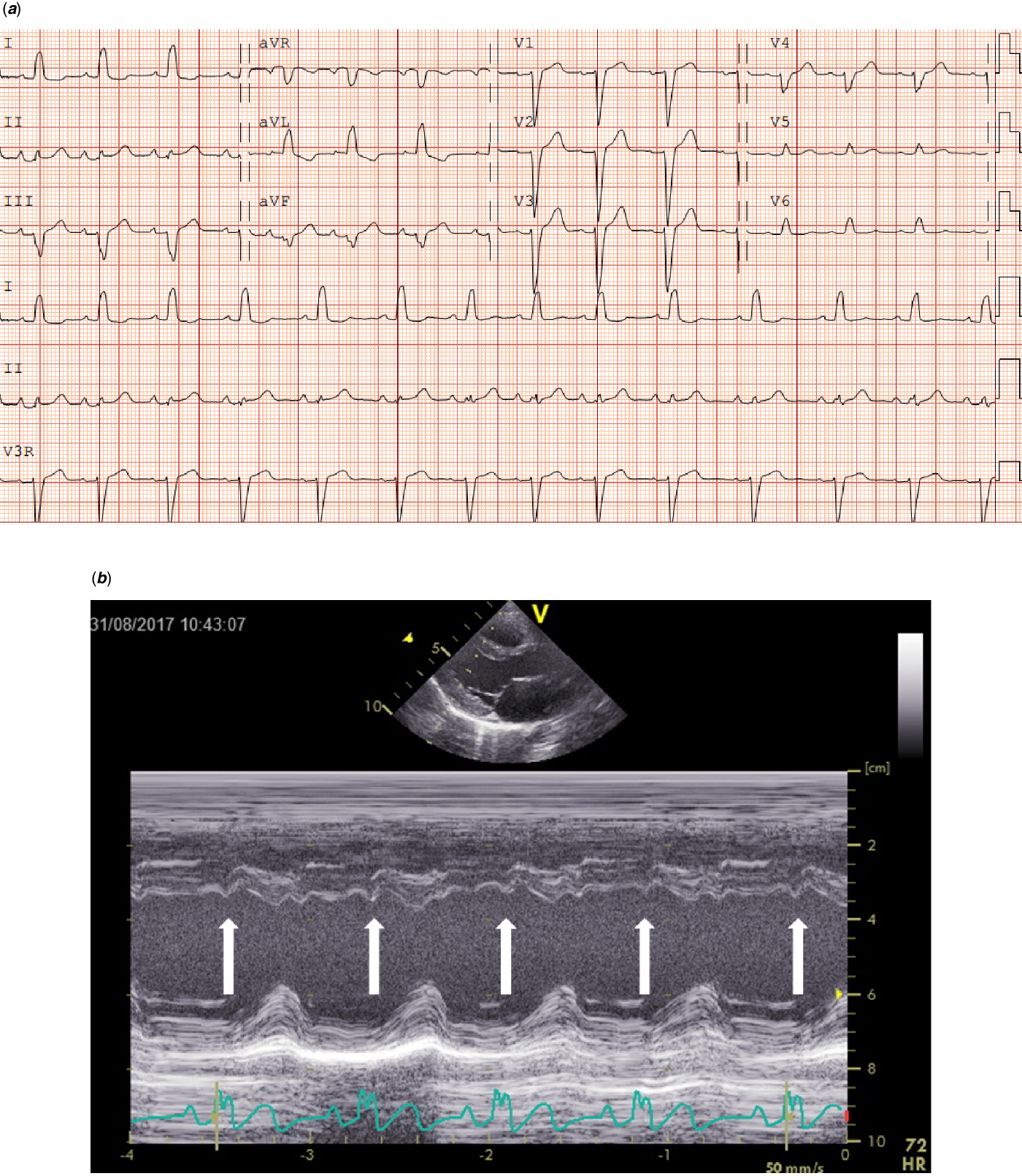
Figure 2. ( a ) Electrocardiogram (speed 25 mm/sec, voltage 0.1 mV/mm) from an 8-year-old girl with a SOS1 mutation with complete left bundle branch block (LBBB). In addition to an duration increase in QRS, please note the distinctive mid-QRS notching in leads I and aVL, along with mid-QRS-slurring in leads V5 and V6. ( b ) Echocardiographic assessment of the septal movement in the long-axis in this patient revealed a septal flash (white arrows), which is a marker of left ventricular electromechanical desynchrony in the presence 'of LBBB.
Table 3. Specific ECG features in patients with different gene mutations without CHDs
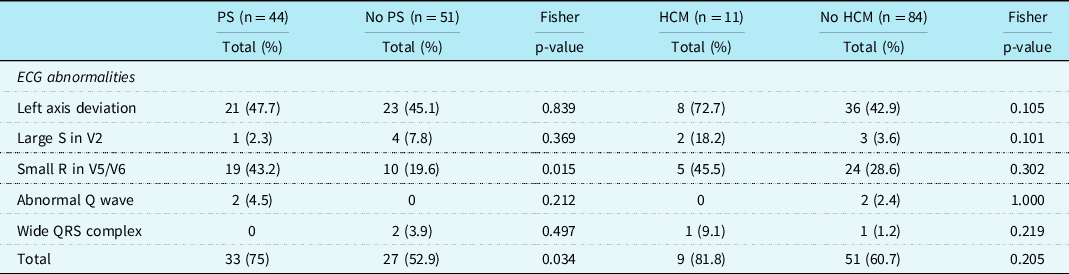
Patients with pulmonary valve stenosis had more often a small left precordial R-wave than patients without a pulmonary valve stenosis(p = 0.015). No other significant differences were found (Table 4).
Table 4. Specific ECG features in patients with Noonan syndrome with and without pulmonary stenosis and with and without hypertrophic cardiomyopathy
Discussion
In this cohort, CHD was found in 62.1% and typical Noonan syndrome-related electrocardiographic features in 63.2% of Noonan syndrome patients. The typical Noonan syndrome-related electrocardiographic features were also present in 41.7% of patients without CHD. Left axis deviation was seen in 46.3% of patients, small left precordial R-waves in 30.5%, and large right precordial S-waves in 5.3% of the Noonan syndrome patients. There was no significant difference in the frequency of the individual- specific Noonan syndrome-related electrocardiographic features between the group with CHD and the group without CHD.
In this retrospective study, the distribution of mutations in PTPN11, KRAS, and BRAF is comparable to those reported in the literature, while the distribution of mutations like SOS1, RAF1, and RIT1 is slightly lower than in some earlier studies. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Yaoita, Niihori and Mizuno9,Reference Ko, Kim, Kim and Yoo17,Reference Zenker, Horn and Wieczorek22–Reference Roberts, Araki and Swanson28 The reported incidence of the CHD found in this study is comparable with previous studies, Reference Kouz, Lissewski and Spranger5,Reference Croonen, van der Burgt, Kapusta and Draaisma7–Reference Allanson, Roberts, Adam, Ardinger, Pagon, Wallace, Bean and Stephens10,Reference Bertola, Kim and Sugayama15 also the high incidence of CHD in patients with a mutation in RIT1 or RAF1. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Kouz, Lissewski and Spranger5,Reference Ko, Kim, Kim and Yoo17 The frequency of pulmonary valve stenosis in patients with PTPN11 and RIT1 gene mutations is the same as found in previous studies, in contrast with patients with SOS1 and BRAF gene mutations, who are found to have less often pulmonary valve stenosis in this study than described in the literature. Reference Lee, Kim, Jin, Kim, Choi and Yoo4,Reference Kouz, Lissewski and Spranger5,Reference Croonen, van der Burgt, Kapusta and Draaisma7 This may probably be due to the small number of patients with SOS1 and BRAF mutations. Except for the small number of patients with a SOS1 mutation, this retrospective study can be seen as a representative for patients with NS.
Compared to the 5% incidence in healthy children and adolescents from a large European cohort we find a much higher incidence of specific electrocardiographic abnormalities in patients with Noonan syndrome. These findings are in line with previous studies of Noonan syndrome. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13,Reference Bertola, Kim and Sugayama15,Reference Molinari, Brunetti and Biasco29–Reference Sanchez-Cascos31 Although there is no significant difference in the incidence of the individual-specific electrocardiographic features between the group with CHD and the group without CHD, specific electrocardiographic features as a group occur significantly more frequent in patients with CHD (p = 0.001). This is in contrast to prior studies, even though the typical Noonan syndrome-related electrocardiographic features are evaluated with the same criteria. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13 This may be due to differences in inclusion criteria of CHD. In recent studies on cardiovascular disease in patients with Noonan syndrome, no patients with valvular regurgitations were described. Reference Jhang, Choi, Lee and Kim32,Reference Pierpont and Digilio33 In our study, we included also patients with mild valvular regurgitations on echocardiogram, where in earlier studies, maybe due to technical possibilities or definitions, some valvular regurgitations may not have been included. Reference Croonen, van der Burgt, Kapusta and Draaisma7,Reference Raaijmakers, Noordam, Noonan, Croonen, van der Burgt and Draaisma13,Reference Pierpont and Digilio33
A part of the specific Noonan syndrome-related electrocardiographic features may be due to the CHD diagnosed. In patients with pulmonary valve stenosis, the baseline electrocardiogram is often normal in cases of mild pulmonary valve stenosis, and in cases with more severe pulmonary valve stenosis, there may be a slight right axis deviation or signs of right ventricular hypertrophy. However, there was no difference between patients with and without pulmonary valve stenosis regarding the specific Noonan syndrome-related electrocardiographic abnormalities (including left axis), except that patients with pulmonary valve stenosis had more often a small R-wave left precordial, which cannot be explained electrophysiologically by the pulmonary valve stenosis. An abnormal electrocardiogram is common in patients with hypertrophic cardiomyopathy. In literature, most often, abnormal repolarization and Q-waves are reported, and/or left axis deviation. Reference Savage, Seides and Clark34,Reference Helmy, Maauof and Shaaban35 However, in our study, left axis deviation or abnormal Q-waves occurred not more often in patients with hypertrophic cardiomyopathy than in patients without hypertrophic cardiomyopathy. We have the experience that the characteristic features (left axis, small R-wave left precordial), do not change in life or after intervention (surgical intervention, trametinib), however, may be somewhat less accentuated (data are not shown).
Previous studies have demonstrated electrocardiographic abnormalities in patients with Noonan syndrome with multiple lentigines (formerly LEOPARD syndrome), mostly reflecting left ventricular hypertrophy. Reference Helmy, Maauof and Shaaban35 More strikingly, a recent study found that non-re-entrant atrial tachycardias, multifocal atrial tachycardia, and ectopic atrial tachycardia can arise from the gain-of-function mutations in multiple genes giving rise to RAS-MAPK pathway dysregulation. Reference Limongelli, Pacileo and Marino36 These cardiac arrhythmias occurred in patients with Noonan and Costello syndrome both in the presence or absence of hypertrophic cardiomyopathy, suggesting that the atrial arrhythmias seen in these patients are not simply caused by the pressure load in these patients. Reference Levin, Saitta and Gripp37 In our study, no serious arrhythmias were seen.
A limitation of this retrospective study is that the echocardiograms were made in a clinical setting, and it was only possible to study 55 of the 95 original recordings. However, all echocardiograms were made according to standard guidelines.
In conclusion, this study seems to be a representative of patients with a genetically proven Noonan syndrome. Specific Noonan syndrome-related electrocardiographic features seen were mainly left axis deviation and small R-waves in the left precordial leads, in patients with or without CHD. Even though some CHD and specific Noonan syndrome-related electrocardiographic abnormalities are significantly more frequent in some gene mutations, the presence is widespread over all mutations. Our results suggest that typical Noonan syndrome-related electrocardiographic features are the one end of the cardiac spectrum and overt CHD the other end.
Acknowledgements
Several authors of this publication are members of the European Reference Network for Developmental Anomalies and Intellectual Disability (ERN-ITHACA).
Author contribution
Eefke Vos: conceptualisation and design of the study, data analysis and interpretation, drafting of the manuscript, approval of the final manuscript. Erika Leenders: conceptualisation and design of the study, critical review of the manuscript, approval of the final manuscript. Sterre R. Werkman: conceptualisation and design of the study, drafting of the manuscript, approval of the final manuscript. Floris E. A. Udink ten Cate: conceptualisation and design of the study, drafting of the manuscript, approval of the final manuscript. Jos M.T. Draaisma: conceptualisation and design of the study, data analysis and interpretation, critical review of the manuscript, approval of the final manuscript.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines on human experimentation (please name) and with the Helsinki Declaration of 1975, as revised in 2008, and has been approved by the Medical Ethics Committee (CMO) of the district Arnhem/Nijmegen (file number 2020-6851).







 Open access
Open access