


Schizophrenia is a devastating, chronic neurodevelopmental disorder that is currently understood as a complex interplay of genetic, epistatic and environmental factors Reference Harrison and Weinberger1,Reference Rapoport, Addington, Frangou and Psych2 that produce a temporally dynamic pattern of illness and progressive structural brain abnormalities. Early-onset schizophrenia, first described by Homburger in 1926, Reference Homburger3 refers to the onset of first sustained positive psychotic symptoms before 18 years of age. 4 Although there is some debate as to true incidence Reference Cannon, Cadenhead, Cornblatt, Woods, Addington and Walker5 by time, place and person, onset of schizophrenia is typically during adolescence or young adulthood.
Generally, the symptoms of adolescent-onset psychosis are similar to those of adult-onset psychosis Reference Schimmelman, Conus, Cotton, McGorry and Lambert6 and diagnosis has been determined using the same criteria (DSM–IV and ICD–104) as adults. However, recent studies of early-onset psychosis suggest that onset in adolescence may be associated with more neurological soft signs and more neuropsychological deficits, Reference Cannon, Cadenhead, Cornblatt, Woods, Addington and Walker5 more negative symptoms Reference Harris, Henry and Harrigan7 and substance use at baseline, Reference Pencer, Addington and Addington8 a lower level of premorbid functioning as well as a longer duration of untreated psychosis than onset in adulthood. Reference Ballageer, Malia, Manchanda, Takhar and Haricharan9 Duration of untreated psychosis has been clearly associated with poorer clinical outcome. Reference Harris, Henry and Harrigan7,Reference Ballageer, Malia, Manchanda, Takhar and Haricharan9
Atypical antipsychotics are often considered the treatment of choice for children and adolescents with schizophrenia. Reference Remschmidt, Schulz, Martin, Warnke and Trott10 However, at the time of initiation of this study in 2000, the efficacious and safe doses of risperidone were unknown. Armenteros assessed the short-term efficacy of risperidone (2–10 mg/day) in ten adolescents (11–18 years) with schizophrenia in an open-label pilot study. Reference Armenteros11 Several case studies had reported efficacy using doses largely consistent with the approved dose range for adults. Reference Balderssarini and Teicher12,Reference Quintana and Keshavan13 There were no data available from double-blind trials or those that examined lower dose ranges in this population.
Risperidone is a benzisoxazole derivative with potent serotonin 5-HT2A and dopamine D2 receptor-blocking properties. In adults, there is clear evidence that risperidone is an effective antipsychotic, ameliorating both positive and negative symptoms of schizophrenia and exhibiting a low rate of extrapyramidal side-effects (EPS). Reference Marder and Meibach14,Reference Chouinard, Jones, Remington, Bloom, Addington and MacEwan15 A risperidone study for first-onset psychosis in adults demonstrated that low doses (1–4 mg/day; modal dose of 3 mg/day) showed similar efficacy to haloperidol in acute symptom amelioration, but better efficacy in the prevention of relapse. Reference Schooler, Rabinowitz, Davidson, Emsley, Harvey and Kopala16 Data from paediatric studies in conduct disorders and autism suggested that doses from 0.25 mg/day to 1.5 mg/day were well tolerated, safe and effective in the treatment of disruptive behavioural symptoms associated with these disorders. Reference Aman, De Smedt, Derivan, Findling and Group17–Reference McCracken, McGough, Shah, Cronin, Hong and Aman20
Given the reluctance to conduct a placebo-controlled trial in an adolescent population with schizophrenia during this time before the Best Pharmaceuticals Children Act and Paediatric Research Equity Act, the best approach was judged to be a dose–response study, having based the higher dose range on the doses shown to be efficacious in adults (1.5–6.0 mg/day) and then using a tenfold lower dose (0.15–0.60 mg/day) as a control. Our study assesses the antipsychotic efficacy, safety and tolerability of two dose regimens of risperidone in adolescents with schizophrenia over an 8-week treatment period. It was postulated that risperidone 1.5–6.0 mg/day would provide statistically significant higher efficacy based on Positive and Negative Syndrome Scale (PANSS) Reference Kay, Fiszbein and Opler21 scores than the lower dose range of risperidone 0.15–0.60 mg/day.
Method
The study was conducted from 24 April 2001 to 13 March 2006 at 41 centres in Belgium (n=6), Bulgaria (n=16), the Czech Republic (n=10), Estonia (n=17), Germany (n=6), Poland (n=92), Romania (n=33) and the USA (n=77).
Participants
Adolescents 13–17 years of age with a DSM–IV diagnosis of schizophrenia, currently hospitalised for an acute episode (PANSS total score between 60 and 120 inclusive) were recruited into the study. A trained child psychiatrist established diagnosis using the Semi-Structured Clinical Interview for DSM–IV for Children of the Kiddie-Schedule for Affective Disorders and Schizophrenia – Present and Lifetime 22 administered individually to parents and children in separate interviews.
The study consisted of a screening phase, during which prohibited psychotropic medications and substances of misuse were washed out for at least 7 days, and an 8-week double-blind treatment phase. If patients worsened during the screening phase, it could be shortened to 3 days with notification to the sponsor. Investigators could administer up to two doses of any psychotropic drug otherwise prohibited by the protocol without requiring a washout period. All patients were hospitalised at study start, after which adequately stabilised patients could be discharged from hospital and continue the study as out-patients. After receiving a complete description of the study, patients' parents or legal guardians signed an informed consent document. The patients also provided written assent. During the consent process, the difference in the two doses was explained to patients and their caregivers. It was explained that the lower dose, although expected to have some activity, might be an ineffective treatment. An independent ethics committee or institutional review board reviewed the study protocol and amendment. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, and was performed consistent with good clinical practices and applicable regulatory requirements.
Female patients of childbearing potential were required to have a negative serum pregnancy test at screening. Sexually active female patients agreed to use adequate contraception throughout the trial. A responsible person was available to accompany the patient to the investigational site to provide information for the PANSS and other evaluations, and to dispense the study medication as directed.
Patients believed by the investigator to be at significant risk for suicidal or violent behaviour during the study were excluded, as were patients with a history of neuroleptic malignant syndrome, tardive dyskinesia, or a known or suspected seizure disorder. Patients with a body mass index (BMI) that was <5th percentile or >95th percentile using the standardised percentile curves for children and adolescents Reference Rosner, Prineas, Loggie and Daniels23 were also excluded to ensure that drug exposure was within defined limits.
Rescue medications for agitation (lorazepam, diazepam, hydroxyzine) or insomnia (zolpidem or zopiclone) were allowed during the washout period and for the first 3 weeks of double-blind treatment.
Antiparkinsonian medications and propranolol were permitted for documented treatment-emergent EPS and akathisia, respectively, after baseline.
Randomization and dosing
Patients were randomised to either risperidone 1.5–6.0 mg/day (regimen A) or risperidone 0.15–0.6 mg/day (regimen B) treatment on day 1 of the double-blind treatment phase using a computer-generated schedule. Risperidone was administered as an oral solution once or twice daily in a divided dose. The starting dose for regimen A was 0.5 mg/day for patients weighing ⩾50 kg or 0.01 mg/kg/day for patients weighing <50 kg (treated with a 1.0 mg/ml solution). The starting dose for regimen B was 0.05 mg/day for patients weighing ⩾50 kg or 0.001 mg/kg/day for patients weighing <50 kg (treated with a 0.1 mg/ml solution). Proposed upward titration schedules were provided for both dose regimens, but investigators were required per protocol to adjust up to the maximum tolerated dose over a period of 12 days to ensure that a full dose range would be explored for safety. The dose was to remain stable during the last 4 weeks of the double-blind period.
The dose range of both arms (in association with psychotherapy and psychoeducation) was believed to provide potential for benefit such that no patient would be deprived of psychiatric treatment and careful clinical monitoring. Additionally, selection criteria were set to exclude high-risk patients, and, although there was a brief washout period at the beginning of the study, risks of exacerbation of symptoms during this period and exposure to subtherapeutic treatment were minimised by allowance of rescue medication and clearly delineated withdrawal criteria.
Assessments
The primary efficacy measure was the baseline-adjusted mean change in the PANSS Reference Kay, Fiszbein and Opler21 total score from baseline to end-point. The PANSS was administered on the basis of a semi-structured interview guide by an adequately trained clinician who did not provide psychotherapy or psychoeducation to the participant. Individual patient ratings were made, when possible, at approximately the same time of day at each visit. If possible, the same person administered the scale at all visits.
To ensure the consistent administration and scoring of the PANSS in the adolescent study population, the sponsor, in consultation with thought leaders, developed and implemented PANSS administration guidelines, a training programme, and an annual certification process for investigators that utilised training tapes with adolescents who showed positive and negative symptoms. The PANSS was translated (and back-translated) to the language(s) of each participating country.
Secondary efficacy measures included:
-
(a) change in total PANSS score from baseline at days 7, 14, 28 and 42
-
(b) change in PANSS factor scores Reference Lindenmayer24 from baseline to end-point
-
(c) the number and percentage of patients achieving a clinical response (i.e. at least 20% improvement compared with baseline at each visit and at end-point on the total PANSS score)
-
(d) change in Clinical Global Impression – Severity Reference Guy25 (CGI–S) score from baseline at each visit and at end-point
-
(e) Clinical Global Impression – Improvement Reference Guy25 (CGI–I) score at each post-baseline visit and at end-point.
In addition, the onset of sustained response (i.e. time until a percentage change in PANSS total score ⩾20% was achieved and was sustained for all subsequent visits) was calculated. Post-hoc analyses also examined the percentage of patients achieving a clinical response of at least 30% and 40% improvement in PANSS total score.
Safety assessments included adverse events (coded using the World Health Organization – Adverse Reaction Terminology (WHOART) dictionary); 26 EPS scales: Abnormal Involuntary Movement Scale, Reference Guy27 Barnes Akathisia Rating Scale Reference Barnes28 and Simpson-Angus Scale; Reference Simpson and Angus29 fasting clinical laboratory testing (including haematology, serum chemistry, prolactin levels and urinalysis); body weight; vital sign measurements; electrocardiograms; urine drug screening; physical examination; and self-assessed Tanner staging. The QT intervals were corrected for heart rate using the Fridericia correction (QTcF) and a linear correction (QTcLD) based on the estimated slope from a regression of QT on heart rate using pretreatment values.
Data analyses
The study was designed so that with approximately 117 evaluable patients (with a baseline efficacy assessment and at least one post-baseline efficacy assessment) per group there would be 90% power to detect a clinically relevant difference of 9 points in change from baseline to end-point between the two groups. The sample size calculation was based on the assumption that the standard deviation of the change from baseline to end-point in PANSS score would be 21.5 points, the standard deviation derived from risperidone studies in adult patients. The intention-to-treat analysis set was defined as all patients randomised (children and adolescents; with schizophrenia or schizophreniform disorder) who took at least one dose of study medication. Because the target population of the study was adolescents with schizophrenia, a modified intention-to-treat analysis set was defined consistent with the criteria outlined in study amendment number two: all randomised adolescents with schizophrenia, age ⩾13 years and ⩽17 years at baseline, who took at least one dose of study medication. Modified intention-to-treat analysis was used for all efficacy analyses and safety summaries. Data from two patients were excluded from the efficacy analyses because of irregularities noted during an audit. The exclusion had no effect on the efficacy conclusions. All statistical tests were two-sided and were interpreted at the 5% level of significance. The last observation carried forward method was applied for missing efficacy data imputation. The change from baseline to end-point in total PANSS score was assessed by an analysis of covariance (ANCOVA) model with treatment and country as the fixed effects and baseline total PANSS as a covariate. Between-group differences in the PANSS-derived response rates were analysed using the Cochrane–Mantel–Haenszel test for general association controlling for country. Changes from baseline in PANSS factor scores and in CGI–S were assessed using a similar ANCOVA model as described above. Between-treatment differences for CGI–I were evaluated using an analysis of variance model with treatment and country as factors. No statistical tests were performed to evaluate between-group differences for any safety parameter.
Results
Demographic and baseline characteristics
A total of 343 patients were screened (Fig. 1). The intention-to-treat analysis set consisted of 279 patients, but 22 of these were excluded from the modified intention-to-treat analyses as they were younger than 12 years at baseline or diagnosed with schizophreniform disorder. Of the 257 patients in the modified intention-to-treat analysis set, 125 received regimen A and 132 received regimen B; 67% of patients in this analysis completed the study (Fig. 1). Mean patient age was 15.6 years (range 13–17). There were slightly more males in the regimen B group; otherwise, demographics were comparable across the dosing groups (online Table DS1). The majority of patients (68%) had a diagnosis of paranoid schizophrenia. Baseline psychiatric characteristics were similar between dose groups (online Table DS1). The mean baseline PANSS total, positive and negative symptoms, and factor scores (Table 1 and online Table DS2) and CGI–S (online Table DS2) scores indicated a population with a marked to severe level of symptoms. In the modified intention-to-treat population, most patients (73%) were receiving medication prior to the study; 68% were treated with psychotropics. Of these, the most common were olanzapine (23%), lorazepam (19%) and haloperidol (13%). Among patients in this analysis set, the median mode dose of risperidone was 0.40 mg/day in the regimen B group and 4.00 mg/day in the regimen A group.
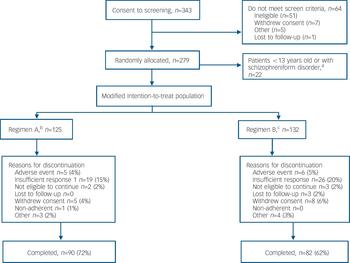
Fig. 1 Flow of participants through the study. a. By Amendment 2 of the protocol, patient age-range changed from ‘2–18 years’ to ‘13–17 years’ and the diagnostic category ‘schizophreniform disorders’ was added to the exclusion criteria. b. Patients ⩾50 kg, risperidone 1.5–6.0 mg/day; patients <50 kg, risperidone 0.03–0.12 mg/kg/day. c. Patients ⩾50 kg, risperidone 0.15–0.6 mg/day; patients <50 kg, risperidone 0.003–0.012 mg/kg/day.
Table 1 PANSS total symptom score: change from baseline to end-point (modified intention-to-treat population) a

| Total PANSS | Regimen A b (n=124) | Regimen B c (n=131) | |
|---|---|---|---|
| Baseline, mean (s.d.) | 96.4 (15.39) | 93.3 (14.14) | |
| End-point, mean (s.d.) | 72.8 (22.52) | 80.8 (24.33) | |
| Change from baseline, mean (s.d.) | -23.6 (22.83) | -12.5 (20.32) | |
| Between-group comparison, change from baseline d | P<0.001 | ||
| Between-group difference (95% CI) e | -10.31 (-15.53 to -5.09) | ||
| Effect size f | 0.49 | ||
PANSS, Positive and Negative Syndrome Scale
a. Data from two participants excluded from analysis because of irregularities noted in a quality assurance audit; exclusion had no effect on the efficacy conclusions
b. Risperidone 1.5-6.0 mg/day
c. Risperidone 0.15-0.60 mg/day
d. Test for no difference between treatment groups from ANCOVA model with factors for treatment group and country and baseline value as covariate
e. Difference (regimen A – regimen B) in least squares means and 95% CI from ANCOVA model
f. Difference in least squares means divided by the square root of the mean square error from the ANCOVA model
Participant disposition
Fewer patients in the regimen A group (n=35, 28%) than in the regimen B group (n=50; 38%) discontinued prematurely from the study for any reason. The most common reason for discontinuation was insufficient response, reported by 19 (15%) patients in the regimen A group v. 26 (20%) patients in the regimen B group (Fig. 1).
Efficacy results
The median mode dose of risperidone (including days off drug) was 4.00 mg/day (range 1.50–6.00) in the regimen A group and 0.40 mg/day (range 0.15–0.60) in the regimen B group. The median duration of risperidone treatment was 56 days in both groups.
About a half of the patients received rescue medications (52%, n=133) for insomnia or agitation during the study. Rescue medication use was higher in the regimen B group.
Statistically and clinically significant improvement in mean total PANSS scores (P<0.001) (Table 1) from baseline to end-point was observed for the regimen A group compared with the regimen B group. The least squares mean change from baseline over time for total PANSS scores (Fig. 2) showed statistically significant superiority of regimen A from the first time point (day 7, P<0.01) through to the end of the study (day 56, P<0.001). Similar results were obtained with an observed case analysis at day 56 showing a difference of 7.5 points on the mean total PANSS score in favour of the regimen A group (P=0.007). The regimen A group had significantly improved compared with the regimen B group on both the positive (P<0.001) and negative (P=0.003) symptom scores (online Table DS2). From baseline to end-point, mean PANSS scores for the other three symptom clusters decreased (improved) in both treatment groups. There was a statistically significant difference indicating greater improvement from baseline in two of the three clusters (P<0.001 for disorganised thoughts; P=0.002 for uncontrolled hostility/excitement) in the regimen A group compared with the regimen B group (online Table DS2). For the anxiety/depression factor, the difference in improvement between groups was not statistically significant (P=0.058) (online Table DS2). The CGI–S score (online Table DS2) improved from baseline to end-point in both groups, but improvement in the regimen A group was significantly greater (P<0.001) compared with the regimen B group. The CGI–I score was significantly lower (P<0.001) in the regimen A group (online Table DS2), confirming greater overall improvement. The percentage of patients with a clinical response (⩾20% improvement) at end-point was significantly higher in the regimen A group (73%) than in the regimen B group (50%, P<0.001). Post hoc analyses of more stringent response criteria showed a similar trend. In the regimen A group, 61% of patients showed at least a 30% improvement in PANSS total scores v. 50% (P<0.001) of patients in the regimen B group, and 43% of patients in the regimen A group showed at least a 40% change in PANNS total scores compared with 30% (P<0.02) in the regimen B group.
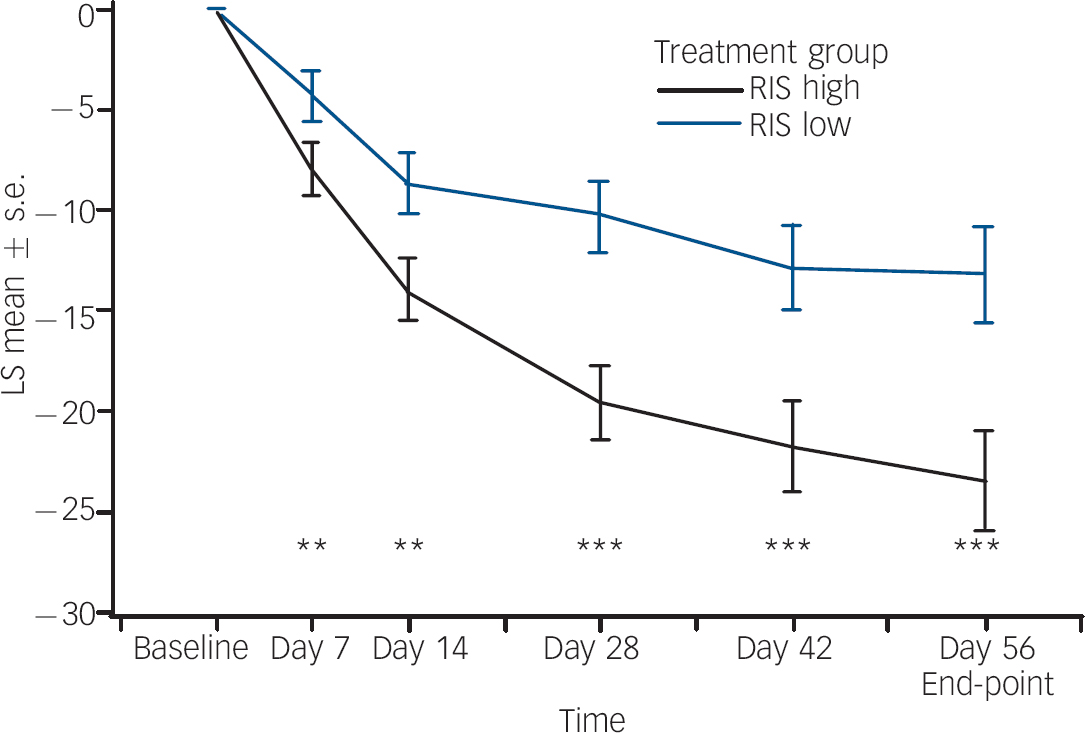
Fig. 2 Least square (LS) mean changes from baseline (and s.e.) in total Positive and Negative Syndrome Scale over time (last observation carried forward, modified intention-to-treat analysis). RIS high, risperidone 1.5–6.0 mg/day; RIS low, risperidone 0.15–0.60 mg/day. ** P<0.01; *** P<0.001.
Sustained response (P<0.001) at the end of the 8-week treatment period was attained in 80 (64.5%) patients in the regimen A group compared with 52 (39.7%) in the regimen B group.
Safety results
Overall, risperidone was well tolerated in this population. Few patients discontinued because of adverse events either in the regimen A (n=5, 4.0%) or regimen B group (n=6, 5.0%). With the exception of treatment-limiting events that were each reported for a single patient (abnormal electrocardiogram, abnormal electroencephalogram and upper respiratory infection in the regimen A group; hypertension, cerebral oedema, tachycardia, increased serum glutamate oxaloacetic transaminase and increased serum glutamate pyruvate transaminase in the regimen B group) the adverse events resulting in treatment discontinuation comprised signs and symptoms associated with the underlying psychiatric disorder.
The proportion of patients experiencing one or more treatment-emergent adverse events was higher among patients who received regimen A (n=93, 74.4%) v. regimen B (n=86, 65.2%) (online Table DS3). In the regimen A group, 24 (19%) patients had their dose adjusted because of an adverse event of somnolence, 8 (6%) because of hypertonia and 8 (6%) because of tremor. No other event led to dose adjustment in more than four patients in the regimen A group. In the regimen B group, four (3%) patients had their dose adjusted because of an adverse event of somnolence. No other event led to dose adjustment in more than two patients this group. Adverse events reported at an incidence at least 5% higher in the regimen A group v. regimen B were: hypertonia, hyperkinesia, somnolence, tremor and weight increase.
Five (4%) patients in the regimen A group and four (3%) patients in the regimen B group experienced serious adverse events, most commonly related to worsening of psychiatric symptoms (reported as ‘psychosis’). There was a single case of cerebral oedema. Two patients in the regimen B group were reported to have had suicidal ideation. There were no suicide attempts reported, although four patients (regimen B, n=3; regimen A, n=1) had events of self-injury and aggression. There were no deaths in the study.
Overall, more patients in the regimen A group (n=41, 33%) experienced EPS-related adverse events compared with the regimen B group (n=13, 10%). The most common EPS-related events were hypertonia, tremor and hyperkinesia (Table 2). None was severe or led to treatment discontinuation. Among patients with EPS-related adverse events, concomitant antiparkinsonian medications were taken transiently by 23 (56%) patients in the regimen A group and by 5 (38%) patients in the regimen B group.
Table 2 Incidence a of treatment-emergent adverse events related to extrapyramidal side-effects

| Adverse event | Regimen A b (n=125), n (%) | Regimen B c (n=132), n (%) |
|---|---|---|
| Dystonia | 23 (18.4) | 8 (6.1) |
| Hypertonia | 18 (14.4) | 6 (4.5) |
| Dystonia | 5 (4.0) | 1 (0.8) |
| Muscle contractions (involuntary) | 1 (0.8) | 2 (1.5) |
| Oculogyric crisis | 1 (0.8) | 0 |
| Tremor | 13 (10.4) | 4 (3.0) |
| Akathisia (hyperkinesia) | 11 (8.8) | 2 (1.5) |
| Dyskinesia | 7 (5.6) | 2 (1.5) |
| Parkinsonism (extrapyramidal disorder) | 5 (4.0) | 0 |
a. Incidence is based on the number of patients experiencing at least one adverse event, not the number of events
b. Risperidone 1.5-6.0 mg/day
c. Risperidone 0.15-0.60 mg/day
There were no clinically relevant changes in vital signs in either group and no patient had an increase of >60 ms in QT interval by either Fredericia (QTcF) or linear-derived (QTcLD) methods of heart-rate correction or for LD at any time point. One female patient experienced an increase from normal baseline QTcLD (433 ms) to a prolonged QTcLD (472 ms) post-baseline.
No clinically significant mean changes in glucose, cholesterol or triglyceride plasma levels were observed. Eleven patients who had glucose values below the upper limit of normal at baseline had values above the upper limit post-baseline. None of these patients had two consecutive fasting glucose measurements above the clinically significant limit, findings considered by the American Diabetes Association as diagnostic for diabetes. 30
Elevations in prolactin levels beyond the upper limit of normal at the end of treatment were observed in 70 (97%) patients in the regimen A group and 55 (64%) patients in the regimen B group. Elevation of serum prolactin is an expected adverse event with risperidone, because of D2 blockade. Elevations in prolactin to levels >100 ng/ml were observed in 45 (18%) patients: 40 females and 4 males in the regimen A group, and 1 female in the regimen B group. Nine patients with elevations in prolactin level above 100 ng/ml experienced potentially prolactin-related events: one male (gynaecomastia) and one female (galactorrhoea) in the regimen B group and five females (three with galactorrhoea, one each with amenorrhoea and hyperprolactinaemia) and two males (one with hyperprolactinaemia and one with breast pain) in the regimen A group. Most of these events occurred after 2 weeks of treatment.
More patients in the regimen A group showed weight gain (82%, n=102) compared with the regimen B group (70%, n=87). The mean change from baseline to end-point in body weight was 3.2 kg (s.d.=3.49) in the regimen A group and 1.7 kg (s.d.=3.29) in the regimen B group. At baseline, about 6% of patients in the regimen B group and 3% in regimen A were at or above the 95th percentile for BMI. Four of 17 patients in the regimen A group and 3 of 25 patients in the regimen B group had shifted from the 85th to <95th (overweight) BMI percentile at baseline to the ⩾95th percentile (obese) at end-point. No patients increased from the <85th percentile to the ⩾95th percentile.
The proportion that experienced weight gain as an adverse event was also higher in the regimen A group (18%, n=22,) compared with the regimen B group (5%, n=7). None of these events was considered serious or led to risperidone discontinuation. In order to assess incremental weight gain in excess of normal growth, z-score analysis was performed using the 30 May 2000 US Centers for Disease Control growth charts (www.cdc.gov/nchs/about/major/nhanes/growthcharts/charts.htm) matched for age. The z-score indicates how many standard deviations an observed value is away from the expected weight or BMI, based on an individual's age and gender. For patients in the regimen A group, the mean baseline weight z-score was 0.21 (s.d.=0.86) and the mean change at end-point was 0.26 (s.d.=0.34). For patients in the regimen B group, the mean baseline weight z-score was 0.35 (s.d.=0.96) and the mean change was 0.10 (s.d.=0.27) (Fig. 3). One half standard deviation from expected age- and gender-matched weight may potentially be used as a proxy for clinically meaningful weight gain, since it compensates for expected change in weight. The end-point change in z-score was >0.5 in 27 (22%) patients in the regimen A group and in 10 (8%) patients in the regimen B group.
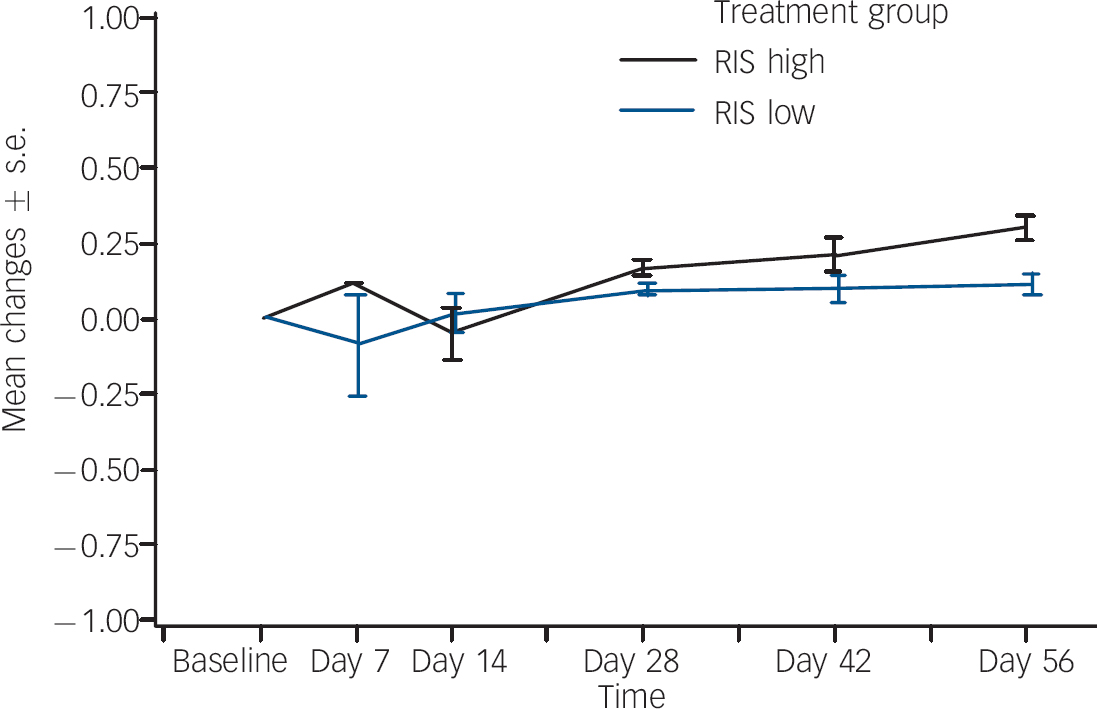
Fig. 3 Body weight z-scores:a mean changes over time. RIS high, risperidone 1.5–6.0 mg/day; RIS low, risperidone 0.15–0.60 mg/day. a. 30 May 2000 US Centers for Disease Control growth charts (www.cdc.gov/nchs/about/major/nhanes/growthcharts/charts.htm).
Discussion
This is the first adequately powered (n=257) controlled study to examine the efficacy, safety and tolerability of two dose regimens of risperidone in adolescent patients with schizophrenia. The higher dose range of risperidone (1.5–6.0 mg/day; regimen A) demonstrated statistically and clinically significantly greater efficacy compared with the lower-dose regimen (0.15–0.6 mg/day; regimen B). Separation in total PANSS scores between the two groups was noted at the first observation, after 1 week of treatment, and was maintained throughout the study. Changes in CGI–S, clinical response and the PANSS factor scores were consistent with the result for the primary end-point, demonstrating that regimen A resulted in statistically and clinically significant improvement compared with regimen B.
Consistent with studies of early-onset psychosis, there was a higher representation of males v. females in this cohort. Symptom severity was marked to severe at presentation, with relatively high negative symptoms scores based on the PANSS negative subset and low functioning based on the CGI–S.
Only one prior published controlled study has examined the effectiveness and tolerability of risperidone in an adolescent population with schizophrenia. Reference Armenteros11 In this earlier study, where very high doses of risperidone were used (mean=6.6 mg/day, range 4–10), response rates were comparable to those reported here for regimen A (mean=4.0 mg/day, range 1.5–6). Recently published open-label data for olanzapine suggest that, although effective, treatment is associated with marked weight gain and laboratory metabolic abnormalities (cholesterol, triglycerides) in adolescent populations. Reference Dittmann, Meyer and Freisleder31 The weight gain observed in our study was more modest.
Both regimens used in this study were well tolerated and the adverse events that occurred were qualitatively similar to those in studies of risperidone treatment in adults Reference Marder, Davis and Chouinard32 and children. Reference Pandina, Aman and Findling33 Additionally, there was a low incidence of treatment discontinuation for adverse events. None of the EPS-related events was severe or led to treatment discontinuation.
Prolactin elevations are commonly observed with the use of D2 receptor antagonists Reference Alfaro, Wudarsky, Nicolson, Gochman, Sporn and Lenane34 in adults and warrant greater evaluation in developing adolescents. In our study, nearly 20% of patients showed elevations in prolactin levels above 100 ng/ml, which were more common in the regimen A than the regimen B group. Only 9 (3.5%) patients experienced potentially prolactin-associated adverse events. In a review of three long-term studies of risperidone treatment for disruptive behaviour disorders in children and adolescents, Reference Pandina, Aman and Findling33 a relatively rapid rise to mean peak prolactin levels above the normal range at 4–6 weeks was followed by a steady decline to mean levels at the upper end of the normal range. Similar patterns were seen in boys and girls. In these studies, few prolactin-related adverse events were observed. In contrast, the emergence of prolactin-related adverse events was higher in adolescent females in our study, which may be attributable to the higher doses utilised. The long-term impact of these changes warrants further study.
Strengths and limitations
This study is the first report on a large population of adolescents with schizophrenia treated with risperidone in a double-blind study. The overall findings complement those from the studies of risperidone monotherapy in adults with schizophrenia. Reference Marder, Davis and Chouinard32 However, several limitations of the study should be noted. First, this study was not placebo controlled. Substantial decreases in PANSS scores compared with baseline were observed in both dose groups. This study, therefore, does not exclude the possibility that the lower doses were also effective. Additionally, because this was not a fixed-dose study and patients were required to be titrated to the maximally tolerated dose within the two dose ranges, interpretations regarding the most effective dose and the reported adverse events must be made cautiously. Results of this study do not allow determination of the optimal effective dose for individual patients. The earliest onset of the antipsychotic effects of risperidone could not be accurately assessed, as efficacy was already significant at the first study time point (day 7). Additionally, a substantial majority of the patients were White, limiting the ability to generalise the results to other ethnic groups. Given that patients believed to be at risk for suicidal or violent behaviour were excluded, caution should be used when extrapolating these results to this sub-population with more severe illness.
Conclusions
Our data suggest that the treatment with risperidone 1.5–6.0 mg/day offers a favourable benefit-to-risk profile for treating adolescents with schizophrenia, a severe psychiatric disorder with potential for long-term disability and with limited treatment options currently available. Improvement was demonstrated across a wide range of specific symptoms, as well as in global functioning, and treatment was associated with acceptable tolerability and safety profiles. Replication of these findings in the presence of a placebo control is warranted.
Acknowledgements
The authors thank Mark Opler (PANSS Institute) for his guidance in the use of the PANSS in this study population; Keith Karcher for additional assistance with statistical analysis and manuscript review; Christine M. Redman for writing and editorial assistance; Wendy P. Battisti for additional editorial assistance; and Sigrid Malbrain and Diane Hoffmann for their assistance in conducting the trial. Drs Redman and Battisti, and Mr Karcher and Ms Hoffman are employed by Johnson & Johnson Pharmaceutical Research & Development, LLC. Dr Malbrain is employed by Johnson & Johnson Research & Development, Division of Janssen Pharmaceutica, NV. The authors also wish to recognise the following main investigators of the 41 sites that enrolled patients. Belgium: Jan Croonenberghs; Bulgaria: Georgui Popov; Svetla Stambolova; Czech Republic: Frantisek Cihak, Ivana Drtilkova, Iva Hodkova, Michal Hrdlicka; Estonia: Evelin Eding, Reet Madisson, A Paaren; Germany: Gottfried Barth, Bernhard Blanz, Elizabeth Grzella, Martin Schmidt, Eberhard Schulz; Poland: Adam Bilikiewicz, Miroslaw Dabkowski, Malgorzata Janas-Kozik, Irena Namyslowska, Jolanta Rabe-Jablonska, Andrzej Rajewski; Romania: Vasile Chirata, Iuliana Dobrescu, Felicia Iftene, Mircea Tiberiu; USA: Scott Aaronson, Eric J. Bartky, Louise M. Beckett, Jeffrey Blumer, Glen Bohlke, Jeffrey Borenstein, Matthew Brams, Kenneth Brown, Stanley Cheren, Ann Childress, James Chou, Jeanette Cueva, Kathleen Degen, Melissa DelBello, Craig Donnelly, Spencer Eth, Fayez El-Gabalawi, Paras Harshawat, Arnold Hartman, Jessica Hellings, Robert Hendren, Scott Hoopes, Jonathan Jensen, Anita Kablinger, Ali Kashfi, Adam Lowy, Paul Markovitz, Kathleen McKenna, Anjali Pathak, Theodore Petti, David Posey, Richard Pugliese, Joachim Raese, Raul Reuben Silva, Robert Riesenberg, Adelaide Robb, Mark Rosato, David Sack, Russell Scheffer, Scott Segal, Lee Stevens, Ekaterina Vassilenko and Bruce Waslick.







eLetters
No eLetters have been published for this article.