



Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disease that presents with varying combinations of behavioral, language, and motor symptoms. Behavioral variant FTD (bvFTD) patients are diagnosed by the presence of apathy, disinhibition, loss of sympathy/empathy, stereotyped behavior, and hyperorality.Reference Rascovsky, Hodges and Knopman1 In addition, bvFTD patients may develop a range of neuropsychiatric symptoms including psychosis, depression, and anxiety.Reference Chakrabarty, Sepehry, Jacova and Hsiung2,Reference Sellami, St-Onge, Poulin and Laforce3 Neuropsychiatric symptoms may precede the onset of cognitive deterioration and lead to diagnosis of a primary psychiatric disorder.Reference Woolley, Khan, Murthy, Miller and Rankin4 FTD patients with predominantly language or motor impairment (motor neuron disease or Parkinsonism) also develop behavioral and neuropsychiatric symptoms.
Due to these protean disease manifestations and the lack of in vivo biomarkers for FTD, it is likely that to date, the prevalence of neuropsychiatric symptoms has been difficult to correctly ascertain. Existing literature on this topic has also been conflicting. A previous review from 2009 found that 6% of reported FTD cases presented with psychosis, and a further 7.2% had psychotic symptoms during the disease course.Reference Velakoulis, Walterfang, Mocellin, Pantelis and McLean5 More recently, a systematic review from 2014 estimated the prevalence of psychosis in FTD at 10–15%.Reference Shinagawa, Nakajima and Plitman6 However, the true prevalence may well be higher, with studies of patients carrying genetic mutations predisposing to FTD showing higher rates, in the range of 21–56% for those carrying the C9orf72 hexanucleotide repeat expansion.Reference Ducharme, Bajestan, Dickerson and Voon7 Earlier reviews included cases that were not confirmed by autopsy, which likely impacts the accuracy of their predictions. FTD patients with psychosis are also more likely to receive psychiatric diagnoses initially, leading to a delay in the diagnosis of dementia and potential exclusion from some of these studies.Reference Woolley, Khan, Murthy, Miller and Rankin4,Reference Ducharme, Dols and Laforce8 Hence, we believe that the true prevalence of psychotic symptoms in FTD remains uncertain.
In recent years, knowledge about the genetic and pathological underpinnings of FTD has rapidly expanded. We, therefore, decided to examine the characteristics of patients with a genetically and/or pathologically confirmed FTD diagnosis who developed psychosis in their clinical presentation and provide an updated review on the prevalence of psychotic symptoms in FTD. Unlike previous reviews, we only selected studies that reported a confirmed genetic or pathological etiology of FTD to improve accuracy of our estimates. This systematic review will also help us better understand the biological basis of psychosis in these patients and outline future research directions based on the identified knowledge gaps.
Method
To identify relevant articles, PubMed, MEDLINE, and Psycinfo databases were searched using the following keywords: “Frontotemporal dementia,” “FTD,” “Frontotemporal lobar degeneration,” “Psychosis,” “Genetics,” “Pathology,” “GRN,” “MAPT,” “C9orf72,” “FUS,” “TDP-43,” and “Tau.” The reference lists of articles of interest were also reviewed to obtain further relevant articles. The literature search returned 289 articles published up to December 2022. A further 27 articles were added through hand search and review of references. After removal of duplicates, the articles were screened with the following inclusion criteria: (1) articles that reported psychosis in FTD patients and (2) articles where genetic and/or pathological data were available for patients (Figure 1). Where studies were published by the same group, they were included unless a study was clearly a subset of a larger, more recent study. The time period of interest for psychosis was the early phase of the illness (i.e. within the first 3–4 years of symptom onset), so this time period was used preferentially when available. FTD had to be diagnosed based on either accepted clinical criteria or neuropathological examination. We also included studies with presymptomatic carriers of known causative mutations for FTD and C9orf72 carriers who were diagnosed with amyotrophic lateral sclerosis (ALS). All variants of FTD were included (behavioral variant FTD, non-fluent primary progressive aphasia (PPA), and semantic variant PPA). Articles that were not written in English were excluded. In articles with incomplete pathological or genetic data for all participants, we also excluded the cases where these data were unavailable.
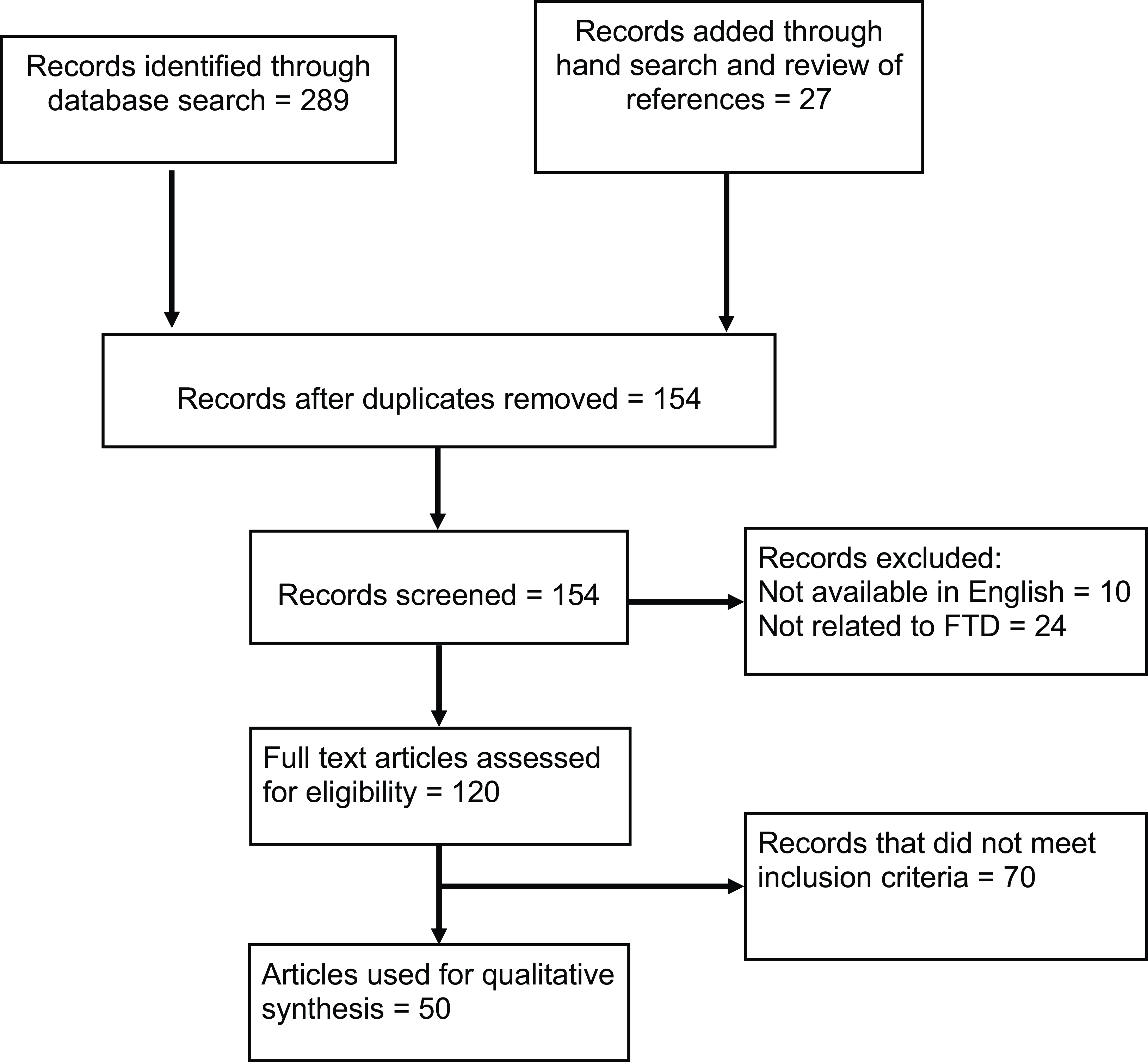
Figure 1: PRISMA flow diagram for systematic literature review.
From these articles, data on psychotic features (delusions and hallucinations), genetic results, and pathological correlations were extracted. The aggregate prevalence of psychosis in FTD patients was calculated from the articles included in our systematic review. The reported prevalence of psychosis in each genetic and pathological subgroup was also calculated. Individual patient data including age of onset, sex, types of psychotic symptoms, and presence of associated features (e.g. ALS) were also extracted wherever available.
Results
Fifty articles met the inclusion and exclusion criteria for this review (Table 1), which comprised 14 case reports, 16 case series, and 20 observational analytical studies (14 cohort studies, 5 case–control studies, and 1 cross-sectional study). Table 1 describes the study design, sample sizes, diagnostic criteria, and analytical methods used in these articles. Thirty-seven articles reported genetic data, 15 articles reported pathological data, and 13 studies reported some combination of genetic and pathological data for the included patients. The studies report a total of 1518 subjects with autopsy-confirmed FTD or carrying genetic mutations associated with FTD, out of which 368 patients (24.2%) developed psychosis (not including single case reports). There were 775 patients with a confirmed genetic mutation, the majority of these carrying the C9orf72 hexanucleotide repeat expansion, and of these patients, 204 (26.3%) developed psychosis. We extracted available clinical characteristics of individual patients with psychosis, which are summarized in Supplementary Table 1 (N = 99).
Table 1: List of articles reviewed for literature synthesis (in order of publication year)
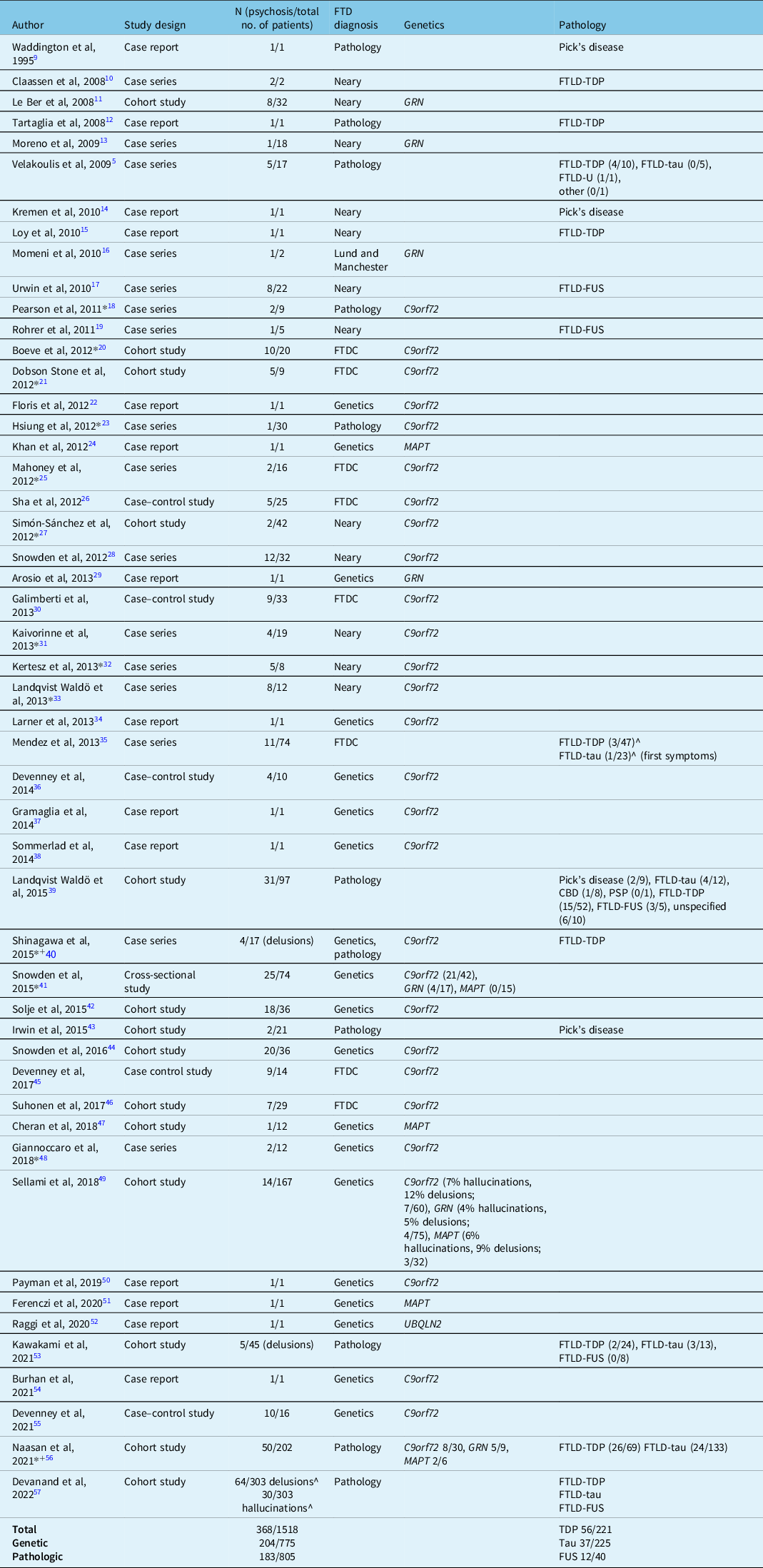
FTDC=international consensus criteria for FTD.
* Study reporting a combination of genetic and pathologic data.
+Study reporting both genetic and pathologic data included in analysis.
^Numbers approximate; taken from graphical representations from the publication.
Genetics
The genetic landscape of FTD has been gradually expanding. At least 30% of FTD patients have a causative genetic mutation, with the C9orf72 hexanucleotide repeat expansion, GRN mutations, and MAPT mutations being the most common.Reference Onyike and Diehl-Schmid58,Reference Greaves and Rohrer59 In addition, mutations in VCP, CHMP2B, TIA1, TBK1, TARDBP, OPTN, and UBQLN2 are reported in small number of families all over the world.Reference Raggi, Bartoletti-Stella, Parchi and Capellari52,Reference Greaves and Rohrer59 Among the rarer genetic mutations, UBQLN2 has been associated with psychosis in a recent case report.Reference Raggi, Bartoletti-Stella, Parchi and Capellari52
C9orf72
A hexanucleotide repeat expansion in the C9orf72 gene, discovered in 2011, is the most frequent genetic cause of FTD and ALS in North America and Europe.Reference Balendra and Isaacs60 This repeat expansion may cause pathogenic effects through multiple mechanisms, including toxic gain-of-function and loss-of-function. According to the putative gain-of-function mechanism, C9orf72 hexanucleotide repeats undergo transcription into repeat RNAs which sequestrate RNA binding proteins and are translated into dipeptide repeats, which form neuronal inclusions.Reference Ducharme, Bajestan, Dickerson and Voon7,Reference Balendra and Isaacs60 According to the putative loss-of-function mechanism, expansion of the hexanucleotide repeat in the promoter region of the C9orf72 gene leads to lower protein levels of C9orf72, which impairs autophagy and lysosomal function.Reference Balendra and Isaacs60
Our literature review revealed 29 articles that reported psychosis in association with the C9orf72 mutation. Out of a total of 557 subjects with the C9orf72 hexanucleotide repeat expansion, 175 subjects (31.4%) developed psychotic symptoms, making it the most common genetic mutation reported in association with psychosis and FTD. Compared to the total number of FTD patients with a confirmed genetic mutation with clinical psychosis we found in the literature (N = 204), C9orf72 mutation carriers constituted the majority (86%), confirming the previously reported assertion that psychosis in the context of a family history of FTD is suggestive of C9orf72 mutation.Reference Snowden, Rollinson and Thompson28 A small study reported that a 10-base pair deletion adjacent to the C9orf72 expansion may be protective against the development of psychosis.Reference Snowden, Harris and Adams44
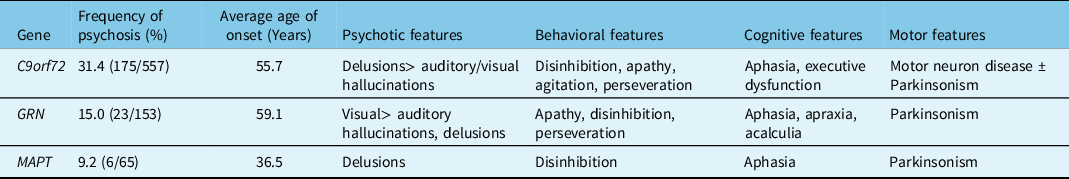
Patients with the C9orf72 hexanucleotide repeat expansion demonstrate wildly different ages of onset and clinical presentations, including bvFTD, FTD-ALS, or ALS alone. The clinical characteristics of C9orf72 carriers are summarized in Table 2. The mean age of onset of symptoms in C9orf72 mutation carriers with psychosis was 55.7 years (Table 2). In addition to behavioral features of FTD, 27.0% of patients developed ALS and 23.6% of patients developed Parkinsonism. Among C9orf72 mutation carriers, delusions were the most common psychotic feature. Patients reported bizarre delusions, hallucinations, or both.
Table 2: Clinical characteristics of FTD patients with a genetic mutation and psychosis
Although most patients with psychosis developed cognitive and behavioral features suggestive of bvFTD, cognitive impairment may trail the onset of psychotic symptoms in C9orf72 mutation carriers.Reference Sommerlad, Lee, Warren and Price38 Patients with longstanding psychosis prior to the diagnosis of bvFTD have also been described.Reference Sellami, St-Onge, Poulin and Laforce3,Reference Kertesz, Ang and Jesso32 In these situations, neuroimaging and genetic testing for the C9orf72 mutation led to the final diagnosis. As patients with the C9orf72 mutation may present with predominantly psychotic features, investigators examined cohorts of patients with a psychiatric diagnosis for the mutation. Screening of two cohorts of patients with schizophrenia and psychosis for C9orf72 mutation did not reveal any mutation carriers, suggesting this is not a common cause of psychosis in the general population.Reference Huey, Nagy and Rodriguez-Murillo61 However, family members of C9orf72 carriers are at increased risk of developing schizophrenia or psychosisReference Devenney, Ahmed, Halliday, Piguet, Kiernan and Hodges62, suggesting some pathogenic role of the C9orf72 mutation in the development of psychosis in sporadic schizophrenia.
GRN
GRN encodes the progranulin protein, which is widely distributed throughout the body and has diverse functions, including neuronal differentiation and anti-inflammatory actions.Reference Hsiung, Feldman, Pagon, Adam, Ardinger and et al.63 The GRN mutation was discovered as an etiology of autosomal dominant FTD in 2006.Reference Baker, Mackenzie and Pickering-Brown64 Since then several pathogenic mutations have been described, which possibly cause neurodegeneration through a loss-of-function mechanism. Heterozygous mutations in GRN lead to haploinsufficiency and loss of functional progranulin protein.
Our literature search identified seven articles that reported psychotic symptoms associated with GRN mutations, which reported a combined prevalence of psychosis of 15.0% among GRN mutation carriers (N = 153). GRN mutations may be associated with bvFTD, a PPA syndrome, or a mixed phenotype.Reference Greaves and Rohrer59 The clinical characteristics of GRN carriers with psychosis are summarized in Table 2. The average age of onset of symptoms in GRN mutation carriers in our literature survey was 59.1 years. Visual hallucinations were the most common psychotic feature in GRN mutation carriers. In addition, cognitive deficits in the form of aphasia, apraxia and acalculia, and motor symptoms of Parkinsonism were present. Analysis of the Genetic FTD Initiative (GENFI) cohort revealed a 4% prevalence of hallucinations and a 5% prevalence of delusions among GRN mutation carriers (N = 75).Reference Sellami, Bocchetta and Masellis49 Several mutations in the GRN gene were reported in patients with psychosis, including deletions (c.753–754del TG) and missense (c.1A > G) mutations. Le Ber et al reported a higher incidence of visual hallucinations among GRN mutation carriers.Reference Le Ber, Camuzat and Hannequin11 They also reported a unique patient who was diagnosed with schizophrenia due to lack of cognitive impairment. Momeni et al also reported a family with the GRN mutation (c.675–676del CA) with history of early-onset psychosis, which was diagnosed as schizophrenia.Reference Momeni, DeTucci and Straub16 A genetic study of families with schizophrenia revealed a significant linkage with the genetic locus 17q21, which contains the GRN gene.Reference Escamilla, Hare and Dassori65
MAPT
The MAPT gene encodes isoforms of the tau protein, which is involved in microtubule stabilization in neurons. MAPT was the first gene associated with the development of FTD, described by Hutton et al in 1998.Reference Hutton, Lendon and Rizzu66 Since then several mutations in the MAPT gene have been described, which cause FTD through different mechanisms, including an altered 3R/4R tau ratio and abnormal tau phosphorylation.Reference Deleon and Miller67
Our literature survey revealed six articles that reported psychosis in association with MAPT mutations. MAPT mutation carriers may present with clinical features of FTD, PPA, or an atypical Parkinsonian syndrome, such as corticobasal syndrome.Reference Greaves and Rohrer59 The articles reported 6 patients with psychotic symptoms out of a total of 65 patients with MAPT mutations (9.2%). The average age of onset of symptoms in patients with psychosis was 36.5 years, though the accuracy of this result is limited by small numbers. In MAPT mutation carriers, delusions were the prevalent psychotic feature, whereas frank hallucinations were rare.Reference Naasan, Shdo and Rodriguez56 Notably, the age of onset of MAPT mutation carriers with psychosis was younger than C9orf72 or GRN mutation carriers, which has also been observed in previous publications.Reference Gasca-Salas, Masellis and Khoo68,Reference Moore, Nicholas and Grossman69 Khan et al described a patient with longstanding psychosis prior to diagnosis of FTD.Reference Khan, Woolley and Chao24 A comparative study reported a lower frequency of psychotic features in MAPT mutation carriers (zero percent) compared to GRN (24% delusions and 6% hallucinations) and C9orf72 (50% delusions and 21% hallucinations) mutation carriers.Reference Snowden, Adams and Harris41 Thirty-three percent of MAPT mutation carriers demonstrated Parkinsonism on neurologic examination.Reference Snowden, Harris and Adams44
Pathology
The pathological cause of clinical FTD is frontotemporal lobar degeneration (FTLD) named after the pattern of brain atrophy identified on macroscopic examination of the brain. FTLD is subclassified based on the type of abnormal protein inclusions observed on histological examination. There are three main types of proteins that accumulate abnormally in FTLD: transactive response DNA-binding protein 43 kDa (TDP-43; FTLD-TDP), tau (FTLD-tau), and the FET family of RNA-binding proteins (FTLD-FET).Reference Neumann and Mackenzie70 C9orf72 and GRN mutations are associated with FTLD-TDP pathology, while MAPT mutations are associated with FTLD-tau pathology. The prevalence of psychosis is different for each pathological class; however, overall 22.7% of cases were found to have psychosis in this review. A single study was found that compared sporadic and genetic cases within pathological classes: it showed that patients with FTLD-TDP type B pathology with and without delusions had a similar percentage that tested positive for the C9orf72 hexanucleotide repeat expansion.Reference Naasan, Shdo and Rodriguez56 This suggests that delusions were not necessarily predictive of the presence of this mutation.
FTLD-TDP
TDP-43 is a widely expressed RNA-binding protein that is predominantly localized in the nucleus of a cell. TDP-43 regulates metabolism of a wide variety of RNA species, which in turn regulate neuronal development and synaptic functions. Fifty percent of FTD patients demonstrate abnormal TDP-43 inclusions in the brain (FTLD-TDP).Reference Deleon and Miller67 Abnormal TDP-43 undergoes posttranslational modifications including phosphorylation and ubiquitination. The normal nuclear staining pattern of TDP-43 is also lost in FTD.
We retrieved 10 articles that reported psychosis in the context of FTLD-TDP pathology from the literature search. The studies reported a combined 223 patients with FTLD-TDP, including single case reports. Overall, 25.3% of patients with FTLD-TDP pathology were reported to have developed psychosis, most of these data taken from any point in the disease course. Among 61 patients with FTLD-TDP pathology and the C9orf72 mutation, 12 patients (19.7%) developed psychosis.Reference Dobson-Stone, Hallupp and Bartley21,Reference Hsiung, DeJesus-Hernandez and Feldman23,Reference Mahoney, Beck and Rohrer25,Reference Landqvist Waldo, Gustafson and Nilsson33 Among 51 patients with FTLD-TDP pathology without the C9orf72 mutation, 21 patients (41.2%) developed psychosis. In psychotic patients with FTLD-TDP pathology, delusions were slightly more common than hallucinations. A wide variety of delusions were reported, including delusion of pregnancy and bizarre somatoform delusions. Two studies reported a high prevalence of hallucinations in the range of 40–50%.Reference Boeve, Boylan and Graff-Radford20,Reference Naasan, Shdo and Rodriguez56 However, multiple studies specifically recruited patients with the C9orf72 mutationReference Pearson, Williams and Majounie18,Reference Dobson-Stone, Hallupp and Bartley21,Reference Hsiung, DeJesus-Hernandez and Feldman23,Reference Mahoney, Beck and Rohrer25,Reference Snowden, Rollinson and Thompson28 , including one of these two studies. Therefore, the studies, which described patients with the C9orf72 mutation, may have influenced the prevalence of psychosis in patients with FTLD-TDP pathology. Furthermore, several studies looked at psychosis at any stage of the disease course, so it could be that they have captured psychosis in late-stage disease, a feature of many neurodegenerative disorders.
Pathologically, the literature does not reveal a clear difference in general histological findings between patients with and without psychosis. Almost all studies reported degeneration of the frontotemporal cortex and variable involvement of the hippocampus, particularly the CA1 subsector.
FTLD-TDP pathology is heterogeneous in its cortical distribution and morphology of inclusions and has been further organized into several pathological subtypes named A to D.Reference Neumann and Mackenzie70 We examined the reported prevalence among these subtypes in patients with psychosis. As some articles did not mention the subtypes specifically, VHR reviewed the pathological description and assigned subtypes according to the current classification. Within FTLD-TDP, subtype B (N = 21) was the most common subtype associated with psychosis in FTD patients, compared to four cases associated with subtype A (N = 4) and one case each with subtypes C and D.
FTLD-Tau
Approximately 40% of patients with FTD demonstrate abnormal tau pathology (FTLD-tau).Reference Neumann and Mackenzie70 Tau is a phosphoprotein that promotes microtubule polymerization and stabilization of microtubules. Different isoforms of tau exist based on splicing of exon 10 of the tau-encoding MAPT gene. The isoforms are labeled 3R or 4R depending on the number of repeats of a 32 amino acid conserved sequence. FTLD-tau is a clinically and pathologically heterogenous group and comprises both 3R tauopathies (e.g., Pick’s disease) and 4R tauopathies (e.g., corticobasal degeneration and progressive supranuclear palsy).
Nine articles described psychosis in association with FTLD-tau pathology in 227 patients, and in total, 16.4% with tau pathology developed psychosis. The average age of disease onset was 59.8 years. Most of the patients with psychosis were from a single study.Reference Naasan, Shdo and Rodriguez56 In this study, hallucinations were slightly more common than delusions, and over 50% of those with psychosis had both delusions and hallucinations. Delusions were less common when compared to patients with FTLD-TDP pathology. In addition, some of the patients with visual hallucinations had concomitant Lewy Body pathology, which may have been a contributing factor.Reference Naasan, Shdo and Rodriguez56 This was supported by logistic regression analysis in another study, demonstrating an increased risk of delusions and hallucinations in patients with any concomitant Alzheimer or Lewy Body pathology.Reference Devanand, Lee, Huey and Goldberg57
Pick’s disease is characterized by argyrophilic, tau-immunoreactive pathological inclusions called Pick bodies, which are predominantly composed of 3R tau.Reference Mackenzie and Neumann71 In this review, 5 articles reported 12 patients with psychosis associated with Pick’s disease, or 17.9% of cases. The mean age of onset of symptoms in psychotic patients with Pick’s disease was 56.3 years. A mixture of delusions and hallucinations were reported in these 12 patients, with the largest study reporting a slightly higher number with delusions than hallucinations, and a small proportion with both.Reference Naasan, Shdo and Rodriguez56 Hallucinations were often auditory.Reference Waddington, Youssef, Farrell and Toland9,Reference Irwin, Brettschneider and McMillan43
FTLD-FUS
In some patients with tau- and TDP-43-negative FTLD pathology, Fused in Sarcoma (FUS) was discovered to be the constituent of pathological inclusions.Reference Urwin, Josephs and Rohrer17 FUS is a member of the FET family of RNA-binding proteins, which also include EWS RNA binding protein 1 (EWSR1) and TAF15.Reference Neumann and Mackenzie70 FUS pathology is also observed in familial ALS, which is a result of mutations in the FUS gene. In contrast, FTD associated with FUS pathology is almost always sporadic.Reference Mann and Snowden72 FUS is involved in several steps of RNA metabolism, which include RNA granule formation, nucleo-cytoplasmic transport, and protein translation. FUS has a low-complexity domain that can form polymers and transition from soluble to insoluble states. This is a putative mechanism for neurodegeneration in FUS pathology.
Patients with FTLD-FUS pathology had a high overall prevalence of psychosis in our literature survey, though the proportion varied in individual studies likely due to small numbers. We retrieved five articles that reported psychosis in the context of FUS pathology.Reference Urwin, Josephs and Rohrer17,Reference Rohrer, Lashley and Holton19,Reference Landqvist Waldo, Gustafson, Passant and Englund39,Reference Kawakami, Arai, Shinagawa, Niizato, Oshima and Ikeda53,Reference Devanand, Lee, Huey and Goldberg57 The prevalence of psychosis was 30% among 40 patients reported in the retrieved articles. Patients with FTLD-FUS pathology had an earlier age of onset compared to sporadic bvFTD, which holds true for patients who develop psychosis as well. Although the ages of onset for all the patients were not available, one study reported three patients with FTLD-FUS and psychosis with an average age of onset 32.7 years.Reference Landqvist Waldo, Gustafson, Passant and Englund39 It was also noted that patients with FTLD-FUS are more likely to receive a psychiatric diagnosis initially due to their early age of onset.
Discussion
We performed a systematic review to examine the relationship between the different genetic and pathological etiologies of FTD and the development of psychosis. Our review demonstrates interesting patterns that point toward biological mechanisms of psychosis. It also highlights areas that need further research to address the current imbalances in the literature. Earlier review articles demonstrated a variable prevalence of psychosis, for instance, Mendez et al observed a wide range in the prevalence of psychotic symptoms from 5 to 23 %,Reference Mendez, Joshi, Tassniyom, Teng and Shapira35 while Shinagawa et al estimated the prevalence of psychosis in FTD as approximately 10%.Reference Shinagawa, Nakajima and Plitman6 Our review with stricter inclusion criteria observed a higher prevalence of psychosis compared to previous reviews (24.2%). Furthermore, we observed that the prevalence of psychosis differed significantly among genetic and pathological groups, which is consistent with previous reports. We have also included new genetic mutations such as the UBQLN2 mutation, in which an association between FTD and psychosis has also been found.
For context, the point prevalence of psychosis in the general population has been estimated at 3.9 per 1000 people (0.39%) and the lifetime prevalence 7.5 per 1000 (0.75%).Reference Moreno-Kustner, Martin and Pastor73 Hence, the prevalence of psychosis in FTD patients greatly exceeds this and cannot be explained by coexisting psychosis from other causes. The prevalence of psychosis in FTD found in our review is also similar to that seen in major depression (estimated at 28%) Reference Jaaskelainen, Juola and Korpela74, but less than that observed in Alzheimer’s disease (estimated at around 41% over the disease course).Reference Ropacki and Jeste75 It is also important to note that the psychotic episodes noted in these cases are a prominent and pervasive clinical feature and are not secondary to delirium.
The highest rate of psychosis in our review was found in patients carrying the C9orf72 hexanucleotide repeat expansion (31.4%). Since the discovery of the C9orf72 mutation as a cause of familial FTD in 2011, the clinical phenotype has been extensively described in several centers. It is the most common mutation reported in FTD patients with psychosis, and the prevalence of psychosis in patients with the C9orf72 mutation is much higher than those with GRN or MAPT mutations. However, in a longitudinal study of neuropsychiatric symptoms in sporadic and genetic FTD patients (GENFI), there was no significant difference in the prevalence of delusions and hallucinations among the genetic groups.Reference Sellami, Bocchetta and Masellis49 This study does include both symptomatic FTD patients and asymptomatic mutation carriers, which may have influenced the prevalence of psychotic symptoms. Longitudinal follow-up of the same cohort found that the prevalence of psychotic symptoms in C9orf72 mutation carriers increases as their disease progresses.
We observed that FTD patients with psychosis associated with different mutations have slightly different clinical profiles. C9orf72 mutation carriers developed prominent, bizarre delusions while GRN mutation carriers developed prominent visual hallucinations. MAPT mutation carriers predominantly reported delusions that became symptomatic at an early age. However, very few MAPT mutation carriers have been reported with psychosis. The predictive ability of the clinical features should be evaluated in larger prospective studies.
At present, it is unclear whether specific structural brain changes in the genetic groups lead to different susceptibility to psychosis. We speculate that each mutation has different effects on different brain networks. In a multi-center study on familial FTD, early changes in the temporal lobe were observed in MAPT mutation carriers, whereas the insula was affected early in GRN mutation carriers, while the thalamus was affected early in C9orf72 mutation carriers.Reference Sellami, Bocchetta and Masellis49
From a pathological perspective, patients with FTLD-TDP and FTLD-FUS pathology are more likely to develop psychosis compared to patients with FTLD-tau. Most of the FTLD-TDP cases with psychosis were associated with TDP-43 pathological subtype B. FTLD-FUS patients with psychosis have an early age of onset, which overlaps with primary psychiatric disorders. Among the different subtypes of FTLD-tau, Pick’s disease patients are slightly more likely to develop psychosis. The reason for the difference in prevalence of psychosis among different pathological groups is not clearly understood. We speculate that this is a result of differential involvement of brain networks by different pathologies. It is noteworthy that both FTLD-TDP type B and FTLD-FUS are the FTLD types located on a spectrum with ALS. Relatives of patients with ALS have higher incidence of neuropsychiatric conditions, including schizophrenia, which is only partially explained by the presence of C9orf72 mutations.Reference Endo, Takashima and Nekooki-Machida76
Our literature search did not reveal any particular pathological distribution associated with psychosis in FTD patients. While the exact neural substrate that leads to psychosis remains unknown, it has been noted that FTD patients with psychosis had higher pathological burden in the right hemisphere.Reference Landqvist Waldo, Gustafson, Passant and Englund39 Another study of patients with ALS and FTD suggests that vulnerability in the thalamo-cortico-cerebellar networks may contribute to this symptom.Reference Devenney, Tu and Caga55 These intriguing observations require corroboration in a larger cohort of patients with all FTLD subtypes. More work is also required to evaluate treatment approaches to psychosis in FTD, which were not explored at any length in the articles reviewed.
The biological basis of psychosis in FTLD could have several causes, including a common localization of FTD-associated neurodegeneration and primary psychiatric disease without apparent neurodegeneration. A provocative recent publication indicated that Disrupted In Schizophrenia-1 (DISC1) protein can coaggregate with TDP-43 inclusions in human FTLD-TDP-43 and in mouse models of TDP-43 FTD.Reference He, Fan, Qi, Yang, Cooper and Zhao77 In the mouse neuroblastoma cell line N2a, knockdown of DISC1 was found to depress activity-dependent dendritic local translation through impairment of protein translation initiation and, in turn, reduced synaptic protein expression, suggesting a link between TDP-43/DISC1 coaggregation and synaptic dysfunction underpinning psychosis. However, there is no association of schizophrenia and DISC1 variants on genome-wide association studiesReference Soares, Carlyle, Bradshaw and Porteous78, despite the original observation that a DISC1 balanced translocation was linked to psychosis in a Scottish family.Reference Benussi, Premi and Gazzina79 The coaggregation of DISC1 with TDP-43, as well as other proteins implicated in FTLD, will require further neuropathological and biological investigations to better understand the implications.
Our current review has several limitations. One important factor is the time course of disease. While many of the genetic studies evaluated the prevalence of psychosis early in the disease course or in the presymptomatic stage, the pathological studies were often retrospective and recorded psychosis at any time throughout the patient’s illness. The prevalence of psychosis tends to rise with increased duration of illness, which was well illustrated across all genetic groups in a recent article from the GENFI group.80 The variable timing of clinical data collection is a significant limitation in this study, which may also explain some of the variability seen in earlier review articles.
Further limitations include the fact that for some of the subgroups analzsed, the absolute number of patients was small and may have been significantly influenced by findings from a single study. The techniques used for gathering data about the presence of psychosis are also variable and include scoring systems such as the Neuropsychiatric Inventory, structured clinical interviews, caregiver reports, and retrospective review of clinical records. This is also likely a source of the heterogeneity found between studies. Finally, our review found a large number of studies reporting psychosis in C9orf72 mutation carriers, which may artificially exaggerate the prevalence of psychosis in FTD patients as a whole.
In conclusion, our systematic review shows a high prevalence of psychosis among genetic and pathologically confirmed cases of FTD. The C9orf72 mutation and its associated TDP-43 pathology as well as FUS pathology have the strongest association with psychosis. However, the current literature may be skewed toward C9orf72 mutation due to its more recent discovery and therefore possibly more comprehensive phenotyping of psychiatric features. Further prospective studies including sporadic and familial cases with neuropathological confirmation will be required to estimate the true risk of psychosis in all FTD patients.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/cjn.2023.248.
Acknowledgments
Dr. Hirsch-Reinshagen acknowledges support from the UBC clinician-investigator program.
Dr. Scott acknowledges support from the VJ Chapman fellowship grant from the New Zealand Neurological Foundation.
Dr. Hsiung acknowledges support from the Ralph Fisher Professorship in dementia research from the Alzheimer Society of British Columbia.
Authors’ contribution
AC and GYRH conceived and performed analysis on this study and drafted the initial manuscript. VHR and NC provided input to the study design, and critical review and revisions on the manuscript. IS updated and revised the manuscript with recent data.
Competing interests
Drs. Chatterjee, Hirsch-Reinshagen, and Scott have nothing to disclose.
Dr. Cashman discloses that he has received grants or contracts from CIHR, Weston Brain Institute, ProMIS Neurosciences, and is on the board of directors of ProMIS Neurosciences, and owns stock and options in ProMIS Neuroscience.
Dr. Hsiung discloses that he has received grants or contracts from CIHR, NIA/NIH, has been a clinical trials investigator supported by Biogen, Cassava, and Lilly, has participated in expert advisory committee supported by Biogen, Roche, and Novo Nordisk, and is the current president of C5R (Consortium of Canadian Centres for Clinical Cognitive Research).





 Open access
Open access