


Historic overview
The study of acid–base equilibrium and its relationship to the diet and disease has been a subject of considerable speculation for at least several centuries. But, before the 19th century, little was known about the concepts of acids and bases, and no means were available to quantify the acid or alkaline load of foods, or of the pH of physiological processes. Not until the second half of the 19th century did nutritional science began to develop, and the chemical components of food start to be analysed. Henry Clapp Sherman's book in 1919, [The] Chemistry of Food and Nutrition, describes the history of nutritional studies performed up to that date (Sherman was the first to quantify nutritional acid load in 1912).
During the next half century, a number of clinical practitioners outside of academia began to observe health improvements in their patients from consuming unprocessed raw fruits and vegetables, instead of the processed food which were increasingly becoming the standard fare, and developed the concept that life is an equilibrium between acid and base. However, it was not until the 1960 s that input–output acid–base balance studies were performed for the first time in healthy adults and patients with chronic renal acidosis(Reference Lennon, Lemann and Litzow1, Reference Goodman, Lemann and Lennon2). There is still a great deal of debate on how to define acids and bases. In the 1980s, Peter Stewart challenged traditional thought and mathematically determined that hydrogen ion and bicarbonate concentrations were dependent variables. He considered the independent variables, which by definition would determine the dependent variables, to be the strong ion difference (the difference in the net charge of cations and anions fully dissociated in solution), the partially dissociated weak acids (albumin, phosphate), and the partial pressure of carbon dioxide (PCO2) of the solution. While a full discussion of the implications of the differences between theories is beyond the scope of the present paper, of significance is the lack of consensus in the fundamental understanding of the mechanisms, as well as the practical applications of acid–base chemistry in physiological systems. A lengthy review published in January 2008 emphasises this point(Reference Kurtz, Kraut and Ornekian3), and excellent reviews of acid–base history are available(Reference Manz4, Reference Story5).
Definitions: acidosis, acidaemia and diet-induced acidosis
For the purposes of the present review, some definitions are in order, though it should be clear that most definitions are not universally accepted. The term acidosis is often used interchangeably with the term acidaemia, with the latter referring to a blood pH of less than 7·35. Correctly used, the term acidosis refers to a process, or a trend toward acidaemia, without necessarily reaching a pH of less than 7·35, or actual acidaemia. The concentration of H+ in blood plasma and various other body solutions is among the most tightly regulated variables in human physiology. Acidosis only becomes acidaemia when compensatory measures to correct it fail. To illustrate the difference between acidosis and acidaemia, take the following example: two processes occurring simultaneously in the same individual, such as a respiratory acidosis combined with a metabolic alkalosis. In this case, if the respiratory trend toward acidosis is greater than the metabolic trend, a pH of less than 7·35 may be reached, and would be considered acidaemia, despite the presence of a metabolic alkalosis. The intensity of each ‘process’ will determine the pH, but the terms themselves (acidosis, alkalosis) do not indicate a certain pH.
What should also be defined, at least broadly, is the net acid load of the diet, the primary topic of the present paper. Diet net acid load can be estimated from measurements of urinary excretion of ammonium, titratable acids and bicarbonate (called net acid excretion; NAE), or can be calculated from dietary constituents (called net endogenous acid production; NEAP). The techniques for measuring or quantifying the acid load of the diet will be discussed in a later section, but, in general, food contributes a net acid or base effect due to the balance between the acid-forming constituents, such as sulfuric acid produced from the catabolism of methionine and cystine in dietary proteins, and the base-forming constituents, for example, bicarbonate, produced from the metabolism of the K salts of organic anions in plant foods. As predicted by the Stewart hypothesis, sodium chloride appears to affect systemic acid–base status independently of the net acid load of the diet, perhaps by affecting renal excretion of Cl− /![]() , or by a strong ion effect(Reference Frassetto, Morris and Sebastian6). The effects of sodium chloride are especially relevant, given the high salt content of the typical diet in industrialised countries.
, or by a strong ion effect(Reference Frassetto, Morris and Sebastian6). The effects of sodium chloride are especially relevant, given the high salt content of the typical diet in industrialised countries.
With an increasing understanding of acid–base chemistry has come recognition of the significant differences between contemporary diets and diets more typical of Homo sapiens ancestors. Although of course we do not know exactly what our hominid ancestors ate, studies in hunter–gatherer tribes suggest a relatively high intake of plant foods compared with modern-day humans(Reference Cordain, Miller and Eaton7). In a recent study estimating the net acid load (NEAP) of 159 hypothetical pre-agricultural diets, 87 % were found to be base producing, with an estimated mean NEAP of negative 88 mEq/d. In comparison, calculations from the US Third National Health and Nutrition Examination Survey (NHANES III) found the average American diet to be acid producing, with an NEAP of positive 48 mEq/d(Reference Sebastian, Frassetto and Sellmeyer8). This represents a switch from the net base-producing diet we ate for the majority of our evolutionary history to the net acid-producing diet we have been eating for only several thousand years. Much of the current research highlights this change and the potential long-term physiological consequences of a chronic low-grade metabolic acidosis among those eating the typical Western diet, and the effects of reducing or eliminating this diet acid load by altering the diet or giving base supplements.
Is acidosis a real physiological phenomenon?
The human body tends to maintain a tightly controlled pH of about 7·40 in the extracellular fluid by respiratory excretion of carbon dioxide and renal excretion of non-carbonic (non-volatile) acid or base(Reference Paulev and Zubieta-Calleja9). Everyday metabolism produces acid as non-volatile sulfate from amino acid catabolism, non-metabolised organic acids and phosphoric and other acids. The kidney reabsorbs all of the filtered bicarbonate (![]() ) and generates new bicarbonate in the collecting duct. Under normal steady-state conditions, the net quantity of acid secreted and the consequent renal generation of new bicarbonate equals the rate of metabolic proton generation, preserving pH balance. In metabolic acidosis, either non-volatile acid accumulates, or
) and generates new bicarbonate in the collecting duct. Under normal steady-state conditions, the net quantity of acid secreted and the consequent renal generation of new bicarbonate equals the rate of metabolic proton generation, preserving pH balance. In metabolic acidosis, either non-volatile acid accumulates, or ![]() is lost (for example, in diarrhoea) and this can be happening even when the plasma
is lost (for example, in diarrhoea) and this can be happening even when the plasma ![]() is within the range considered to be normal (24–28 mmol/l)(Reference Gluck10).
is within the range considered to be normal (24–28 mmol/l)(Reference Gluck10).
While acute acid loading may only temporarily disrupt acid–base equilibrium, a chronic perturbation occurs when metabolism of the diet repeatedly releases non-carbonic acids into the systemic circulation in amounts that exceed the amount of base released concomitantly (for example, bicarbonate from combustion of organic acid salts of K in vegetable foods)(Reference Frassetto, Todd and Morris11). The size of the discrepancy between acid and base production determines the NEAP rate. To maintain equilibrium when there is a net retention of acid (H+), at least three compensatory physiological responses are activated: buffering, increased ventilation, and increased renal reabsorption and generation of ![]() . The major reservoir of base is the skeleton (in the form of alkaline salts of Ca) which provides the buffer needed to maintain blood pH and plasma bicarbonate concentrations. To some degree, skeletal muscle also acts as a buffer(Reference Bailey, England and Long12). Respiratory ventilation increases within minutes if the acidosis is great enough, and the kidneys compensate by increasing
. The major reservoir of base is the skeleton (in the form of alkaline salts of Ca) which provides the buffer needed to maintain blood pH and plasma bicarbonate concentrations. To some degree, skeletal muscle also acts as a buffer(Reference Bailey, England and Long12). Respiratory ventilation increases within minutes if the acidosis is great enough, and the kidneys compensate by increasing ![]() reabsorption, H+ secretion, and production of the urinary buffer ammonia, all in response to an acidic load.
reabsorption, H+ secretion, and production of the urinary buffer ammonia, all in response to an acidic load.
Thus the loss of bone mass due to acidosis has generally been considered a passive process, a physico-chemical dissolution of the matrix. However, bone dissolution is more than just a passive process, but rather is one of active resorption by osteoclasts, with extracellular H+ being a key inducer of osteoclastic activity. Indeed, extracellular H+ has been suggested to be the ‘long-sought osteoclast activation factor’, partly because in vitro experiments show osteoclasts to be almost inactive at a pH above 7·4, with pH reductions of ≤ 0·1 sufficient to cause a doubling of resorption pit formation, removing both mineral and organic components of bone(Reference Arnett13). A number of in vitro studies have shown that even very small changes in pH have an effect especially on osteoclastic resorptive activity and on osteoblastic activity, with acidosis exerting a selective, inhibitory action on matrix mineralisation(Reference Brandao-Burch, Utting and Orriss14–Reference Bushinsky16). Most recently, Frick et al. have demonstrated that ovarian cancer G protein-coupled receptor-1 (OGR1) is the proton-sensing receptor on the osteoblast that leads to osteoclast activation(Reference Frick, Krieger and Nehrke17).
In vivo studies have generally supported the in vitro findings that acid-promoting diets are associated with both increased Ca and increased bone matrix protein excretion (used as a marker for estimating bone loss), and that neutralising the acid intake with diet or bicarbonate supplements decreases urine Ca and bone matrix protein excretion(Reference Buclin, Cosma and Appenzeller18–Reference Sebastian, Harris and Ottaway21). In a trial of 170 postmenopausal women, for example, potassium bicarbonate supplementation reduced daily urinary Ca excretion, and one could predict which women would benefit most – those with the greatest urinary Ca loss(Reference Frassetto, Morris and Sebastian22).
Additionally, the same mechanism may be involved in Ca nephrolithiasis(Reference Osther23–Reference Osther, Bollerslev and Hansen25). A common risk factor for Ca stone formation appears to be hypocitraturia, which has been associated with a low urinary K level and a more acidic urinary pH, both of which can be predicted by dietary intake(Reference Domrongkitchaiporn, Stitchantrakul and Kochakarn26, Reference Borghi, Schianchi and Meschi27). However, it appears that dietary acid load is a better predictor of urinary citrate than the intake of most individual nutrients, including dietary K. In a trial of 187 patients with renal Ca stone disease, an inverse correlation ( − 0·18) was found between daily potential renal acid load (PRAL; defined below) and daily urinary citrate (P < 0·01). PRAL was also inversely related to urinary pH and K, and although dietary protein and K were correlated with citrate excretion, these relationships did not reach statistical significance(Reference Trinchieri, Lizzano and Marchesotti28). A low-salt, low-animal protein diet has also been shown to reduce risk of stone recurrence (relative risk 0·49) with men followed over 5 years(Reference Borghi, Schianchi and Meschi27). While there are several possible explanations for this effect, one is that the reduced salt and/or animal protein diet is likely to result in a lower acid load.
Finally, metabolic acidaemia has been associated with increased protein breakdown in rats(Reference Mitch, Medina and Grieber29) and possibly subjects with end-stage renal disease(Reference Papadoyannakis, Stefanidis and McGeown30). Mitch et al. demonstrated that increased activation of a ubiquitin–proteasome pathway (via an insulin- or insulin-like growth factor-1-mediated transporter(Reference Du and Mitch31)) increases the production of amino acids which are then made into glutamate in the liver, transported to the proximal renal tubular cells, and then excreted as ammonia (NH3) and complexed to H+(Reference Mitch, Medina and Grieber29). This mechanism allows the kidneys to dramatically increase the amount of acid excreted daily.
What are the causes of acidosis?
The causes of metabolic acidosis include increased consumption or generation of organic acids, as well as either insufficient production of bicarbonate, or renal and/or gastrointestinal loss of bicarbonate, such as that seen in renal disease, diarrhoea, pancreatic drainage and biliary fistula. Causes of renal tubular acidosis include Sjögren's syndrome, systemic lupus erythematosus, urinary tract obstruction, fever, aldosterone deficiency and glucocorticoid administration(Reference Gluck10). Many of these medical conditions are typically associated with overt laboratory abnormalities; either a frank acidaemia, a decrease in plasma bicarbonate, or an increase in the anion gap.
In comparison, diet-induced ‘low-grade’ metabolic acidosis has only very small decreases in blood pH and plasma bicarbonate within the range considered to be normal. Within that range, this means that the system equilibrates nearer the lower end of normal rather than the higher end of normal (see Fig. 1). But, if the duration of the acidosis is prolonged or chronically present, even a low degree of acidosis becomes significant. This less severe but more chronic ‘low-grade’ acidosis is thought to be brought about primarily by two factors: advancing age with a consequent decline in renal function, and diet, which may promote acidosis by both its net acid load, as well as its sodium chloride content. With age, the severity of diet-dependent acidosis increases independently of the diet, most likely due to a decline in kidney functional capacity with age(Reference Frassetto, Morris and Sellmeyer20, Reference Du and Mitch32, Reference Frassetto, Morris and Sebastian33). Renal insufficiency contributes to a metabolic acidosis by reducing conservation of filtered bicarbonate and excretion of acid.
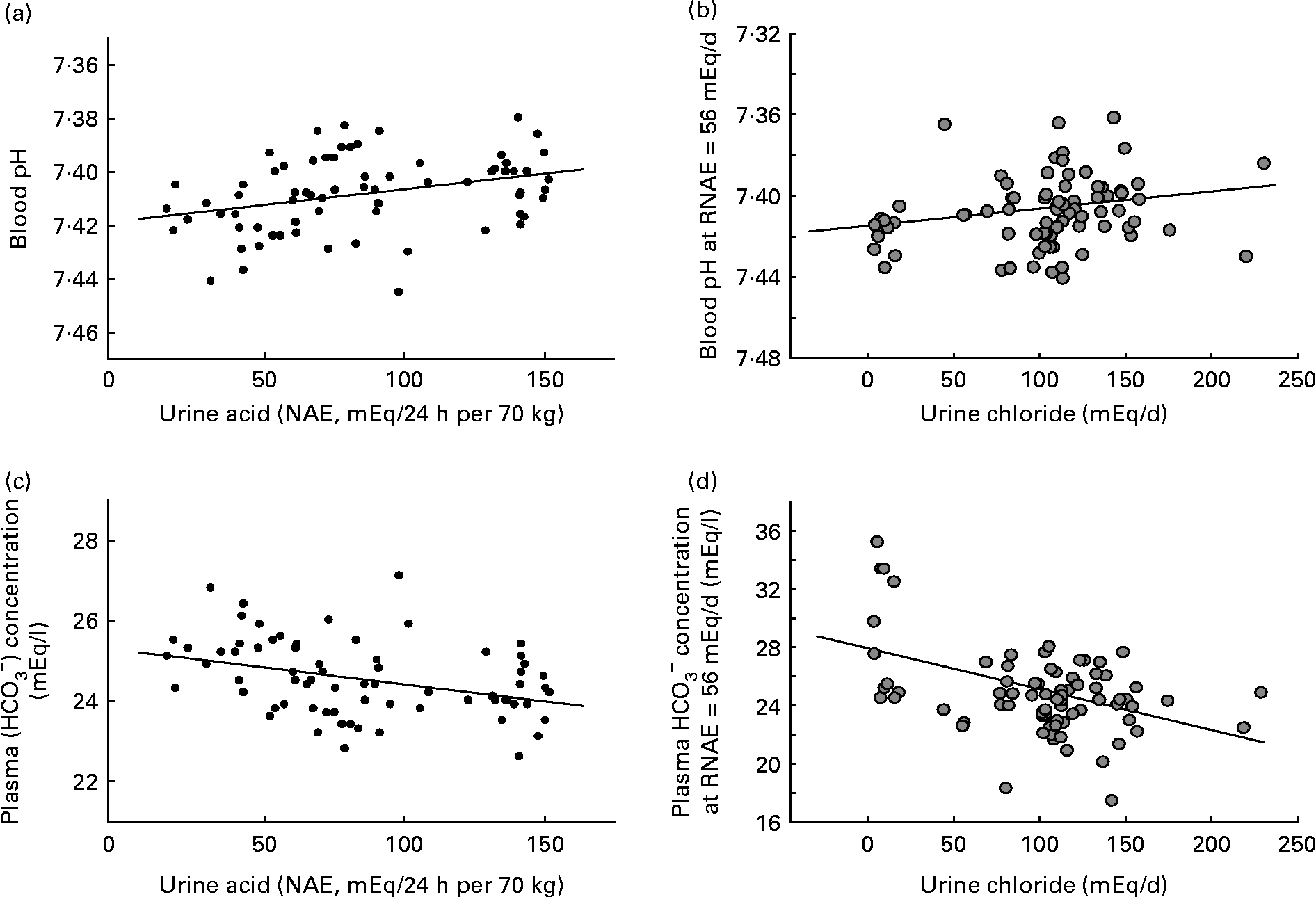
Fig. 1 (a) Blood pH at constant net acid excretion (NAE) rate (r 0·34; P < 0·005). (b) Blood pH at constant urinary chloride excretion rate (renal NAE (RNAE) = 56 mEq/d) (r 0·22; P < 0·05). (c) Plasma bicarbonate at constant NAE rate (r − 0·36; P < 0·005). (d) Plasma bicarbonate at constant urinary chloride excretion rate (RNAE = 56 mEq/d) (r − 0·45; P < 0·001). Adapted from Frassetto et al. (Reference Frassetto, Morris and Sebastian6).
Diet's contribution to an acidotic state is now well documented. In the European Prospective Investigation into Cancer and Nutrition (EPIC) study of over 22 000 men and women, after adjusting for age, BMI, physical activity and smoking, a more alkaline diet (as calculated by PRAL (a diet-only-based estimate of the production of endogenous acid) was significantly associated with a more alkaline urine, both before and after adjustment for age, physical activity and smoking, and after exclusion for urinary protein, glucose and ketones, and for those diagnosed with high blood pressure and/or diuretic medication use (all factors known to influence urine pH)(Reference Frassetto, Morris and Sebastian34).
How is acidosis diagnosed?
Several approaches have been used to estimate the acid–base equilibrium of the body, as well as the net acid contribution from the diet. As discussed previously, this is an area without general consensus, and one that lacks a definitive marker of acid–base status. Because plasma pH is so tightly regulated, without acidaemia an acidosis may not be detected, despite the stress on the body's buffering capacity. Additionally, it has been shown that plasma bicarbonate level may be normal even when acidosis is present, and so neither plasma bicarbonate nor the serum anion gap is a sensitive indicator of acidosis(Reference Gluck10, Reference Frassetto and Sebastian35). This situation is analogous to the impaired glucose tolerance that precedes frank blood sugar elevation. Waiting for acidaemia before recognising acidosis is not a sound clinical strategy.
NEAP represents the amount of net acid (acid minus base) produced by the metabolic system every day (i.e. a combination of cellular metabolism and exogenous acid and base loads from the diet). NEAP can either be measured by quantifying the inorganic constituents of diet, urine and stool, and of the total organic anion content of the urine (calculated as the sum of urinary inorganic sulfate and organic acid salts minus dietary organic anions less faecal organic anions)(Reference Lennon, Lemann and Litzow1), or from estimations of acid or base production from the constituents of the diet. Several methodologies have been proposed to estimate NEAP from dietary components(Reference Frassetto, Todd and Morris11, Reference Welch, Mulligan and Bingham36,37). To avoid confusion, when the NEAP is estimated (which it usually is), it is identified as ‘estimated NEAP’, and the algorithm used for the estimation clearly specified (see Table 1)(Reference Remer and Manz38).
Table 1 Algorithms to determine net endogenous acid production (NEAP)

PRAL, potential renal acid load; OAest, organic acids estimated; Pi, inorganic P; TA, titratable.
The net amount of acid produced daily (NEAP) is closely tied to the net amount of acid excreted (NAE) by the kidneys daily. NAE can also be measured by quantifying the amount of acids in the form of ammonium and titratable acid in the urine and subtracting the amount of base (bicarbonate)(Reference Frassetto, Morris and Sebastian34). When measured directly, nearly 90 % of the variance in renal NAE among subjects is accounted for by differences in NEAP, and because of this closeness in value, they are often considered equivalent(Reference Frassetto, Morris and Sellmeyer20).
Another test often used to estimate the NEAP is the 24 h urine pH. Urinary pH represents an index of the diet-dependent NAE (correlation with calculated NAE, r 0·83; P < 0·001)(Reference Remer, Dimitriou and Manz39), as well as the PRAL(Reference Frassetto, Morris and Sebastian34, Reference Frassetto, Lanham-New and Macdonald40). Additionally, urine pH can be adjusted to a target pH based on PRAL calculations for dietary intake. However, pH strip measurement of the first voided urine was not found to be predictive of the NEAP reflected by the 24 h urine NAE(Reference Remer and Manz41).
Lastly, while urinary Ca is not considered a specific marker for acid–base equilibrium, it has been used as a marker of bone turnover, and elevated levels are a risk factor for kidney stone formation(Reference Michaud, Troiano and Subar42–Reference Pietschmann, Breslau and Pak45). As discussed below, it does not necessarily indicate increased bone Ca loss, however. It could, for example, represent a change in intestinal absorption.
In summary, analysed NAE appears to be the closest approximation of ‘true’ NEAP, and is more easily determined. For simplicity, however, various computational models for estimating NEAP are often employed, each with its own advantages and disadvantages (see Table 1). For a more complete discussion of the pros and cons for each methodology, the reader is referred to a recent review(Reference Stitchantrakul, Kochakarn and Ruangraksa46).
How is acidosis normalised?
The normalisation of a low-grade chronic metabolic acidosis has been accomplished by two methods: change in dietary patterns and alkaline supplementation. Dietary factors that affect net acid production include the quantity and type of protein intake, fruits and vegetables and table salt (sodium chloride). Alkali supplementation is generally in the form of potassium or sodium bicarbonate or citrate.
Increased fruit and vegetable consumption, as well as K and Mg alkali intake, is consistently associated with a base-producing diet(Reference Damasio, Massarino and Durand47). It has been shown that a vegetarian diet has a considerably lower NEAP than both a high and moderate omnivorous protein intake(Reference Welch, Mulligan and Bingham36).
Reducing protein consumption down to the US dietary recommended intake in a trial of thirty-nine healthy premenopausal women has also been shown to reduce Ca excretion and raise urinary pH, as well as reduce markers of bone resorption(Reference Frassetto, Morris and Sebastian48). It should be emphasised that this trial did not evaluate a low-protein diet, but rather lowered what could be considered a high protein intake to a level of 0·8 g/kg for this population. Because renal ![]() formation is dependent upon adequate protein intake, an extremely protein-deficient diet may also increase acidosis(Reference Tucker, Hannan and Chen49). In fact, in a recent study of 161 postmenopausal women, protein intake had a positive association with lumbar bone mineral density, but only after adjusting for the negative effect of the sulfur content of the protein (sulfate), perhaps ‘reconciling reports of positive impacts of dietary protein on bone health with reports of a negative impact of the acid load from sulfur-containing amino acids’(Reference Ince, Anderson and Neer50). In children, a greater protein intake has been associated with greater bone strength, though this effect is negated if alkalinising nutrients are lacking. It should be noted, however, that clearly bone may be influenced by these minerals in ways unrelated to acid–base chemistry(Reference Remer51).
formation is dependent upon adequate protein intake, an extremely protein-deficient diet may also increase acidosis(Reference Tucker, Hannan and Chen49). In fact, in a recent study of 161 postmenopausal women, protein intake had a positive association with lumbar bone mineral density, but only after adjusting for the negative effect of the sulfur content of the protein (sulfate), perhaps ‘reconciling reports of positive impacts of dietary protein on bone health with reports of a negative impact of the acid load from sulfur-containing amino acids’(Reference Ince, Anderson and Neer50). In children, a greater protein intake has been associated with greater bone strength, though this effect is negated if alkalinising nutrients are lacking. It should be noted, however, that clearly bone may be influenced by these minerals in ways unrelated to acid–base chemistry(Reference Remer51).
Finally, increasing sodium chloride intake dose-dependently decreases blood pH and plasma bicarbonate levels(Reference Thorpe, Mojtahedi and Chapman-Novakofski52), independent of the partial pressure of carbon dioxide (PCO2), creatinine clearance and dietary acid load(Reference Frassetto, Morris and Sebastian6). This effect may be due to a decrease in the strong ion difference, as total chloride concentration increases relative to total Na concentration, an effect that may increase H+ concentration(Reference Alexy, Remer and Manz53). Subjects who are particularly sensitive to salt, generally defined as an increase of 3 to 5 mmHg for a given salt load, have more of a metabolic acidosis than those subjects who are salt resistant(Reference Frings-Meuthen, Baecker and Heer54). So, while everyone's net acid load would improve by lowering their dietary salt intake, some individuals should benefit more than others from this dietary intervention.
A number of supplemental interventions have also been used. Salts of carbonic acid are available in a variety of formats. These include sodium or potassium bicarbonate and calcium carbonate. Alkali salts are also available as citrate, acetate or hydroxides. As suggested above, giving Na salts may be partly counterproductive, given their other effects, and so most studies use K or Ca alkali salts. These salts dose-dependently decrease NAE(Reference Kellum55, Reference Sharma, Kribben and Schattenfroh56).
Caution using alkali therapy without careful consideration and expertise in subjects with heart, lung or kidney disease is needed. In congestive heart failure, sodium bicarbonate impairs arterial oxygenation and reduces systemic and myocardial oxygen consumption in these patients, which may lead to transient myocardial ischaemia(Reference Lemann, Gray and Pleuss57). Additionally there may be several simultaneous processes affecting acid–base status among patients with congestive heart failure(Reference Frassetto, Morris, Sebastian, Burckhardt and Dawson-Hughes58). Similarly, bicarbonate loading may worsen exercise response in chronic obstructive pulmonary disease patients(Reference Bersin, Chatterjee and Arieff59). Finally, subjects with kidney failure may develop elevated blood K levels and potentially fatal cardiac arrhythmias if given K alkali salts, or volume overload and breathing problems if given Na alkali salts.
Is acidosis clinically relevant?
Finally, from a clinical standpoint, the most important question is whether or not acidosis has pathophysiological effects that could be ameliorated or abolished by reversing the acidosis. These detrimental effects are associated with long-term acidosis (years to decades), and offer significant potential for prevention-based interventions. Areas of recent interest on the effects of chronic low-grade metabolic acidosis include effects on bone, muscle and kidney stone formation. While the data are generally consistent for the effects of alkali salts on urine Ca, bone biomarker or urinary N excretion (that is, shorter-term effects), the data are less consistent for the longer-term effects of alkali salts on bone mineral density, muscle function or fracture incidence. Positive correlations have been demonstrated using epidemiological or cohort studies, but results of prospective, randomised, placebo-controlled studies on long-term outcomes are mixed.
Bone
In an examination of over 1000 women between the ages of 45 and 54 years, a lower dietary intake of acid-producing foods correlated with greater spine and hip bone mineral density, as well as greater forearm bone mass, after adjusting for age, weight, height and menstrual status(Reference Frangiosa, De Santo and Anastasio60). In the Study of Osteoporotic Fractures Research cohort, over 1000 women aged 65+ years were enrolled in a prospective cohort study. Those with a high dietary ratio of animal to vegetable protein intake (a marker for a greater NEAP) were found to have more rapid femoral neck bone loss and a greater risk of hip fracture than did those with a low ratio(Reference Coppoolse, Barstow and Stringer61).
A number of trials have shown that the bone loss can be reversed by the addition of a base(Reference Sebastian, Harris and Ottaway21). Potassium bicarbonate has been shown to improve Ca and P balance, reduce bone resorption rates, and mitigate the normally occurring age-related decline in growth hormone secretion(Reference Frassetto, Morris and Sellmeyer20). Potassium citrate combined with calcium citrate may be more beneficial than either alone, as demonstrated in a cross-over trial on bone turnover in postmenopausal women(Reference New, MacDonald and Campbell62). Urinary Ca excretion and markers of bone health were improved with potassium citrate, more so in those consuming a high-Na diet(Reference Sellmeyer, Stone and Sebastian63). A recent randomised cross-over trial enrolled postmenopausal women, and introduced four well-controlled diets with varying amounts of calcium and sodium chloride, and evaluated bone Ca balance. They found that at low Ca intakes, the bone Ca balance was negative at varying degrees of sodium chloride intake, but at high Ca intakes, a positive balance was only seen when Na intake was low(Reference Sakhaee, Maalouf and Abrams64). Similarly, in a trial of men with step-wise increases in sodium chloride intake, indications of increased bone resorption and metabolic acidosis were seen with increasing salt intake, and a change in blood pH change averaging approximately 0·02(Reference Thorpe, Mojtahedi and Chapman-Novakofski52).
In a prospective, controlled, trial, 109 men and women with kidney stones had a bone mass increase by increasing their daily alkali intake (potassium citrate), an effect attributed directly to alkalinisation because urinary Ca excretion did not change(Reference Sellmeyer, Schloetter and Sebastian65). Another prospective, blinded study using potassium citrate in 161 postmenopausal women also demonstrated an increase in bone mass over a 12-month period. The authors concluded that ‘as a proof-of-principle study, it demonstrates that neutralization of diet-induced endogenous acid production increases BMD (bone mineral density), thereby proving the concept that such dietary acid loads are detrimental to bone mass and thus constitute a causative risk factor for bone loss in postmenopausal women with osteopenia’(Reference Teucher, Dainty and Spinks66).
On the other hand, a recent prospective randomised placebo-controlled trial by MacDonald et al. did not demonstrate a difference in bone mineral density by dual-energy X-ray absorptiometry after 2 years of potassium citrate supplementation of either 55·5 or 18·5 mEq/d, or an increase in fruit and vegetable intake by 300 g compared with placebo in 276 postmenopausal women(Reference Vescini, Buffa and La Manna67). Interestingly though, they found a significant correlation between the amount of fruit eaten at baseline and bone mineral density at the hip in the entire cohort. And the authors report that ‘food diaries and checklists suggested good compliance for most women, but the blood measurements [plasma vitamin C, homocysteine, and whole cell folate] did not corroborate this’.
A recent study suggests that bicarbonate has favourable effects on bone resorption and Ca excretion compared with K (as potassium chloride), which calls for greater clarification as to the active component in supplemental therapies(Reference Jehle, Zanetti and Muser68). Unless Ca-balance studies are performed, rather than just urinary Ca measurements, it cannot be certain that lower urinary Ca or a decline in bone markers actually translates into less bone loss. For example, decreased urinary Ca may be due to reduced Ca absorption and not due to less bone loss. This point is emphasised in a recent meta-analysis (Fenton et al. (Reference Macdonald, Black and Aucott69)) which found that although there is a linear association between urinary Ca excretion in response to the changes in NAE, this does not necessarily mean that the increased Ca in the urine is from bone. However, they did find that over 20 years, the quantity of excess urinary Ca is consistent with the loss of almost half of skeletal Ca and severe osteoporosis, though their study did not show causality(Reference Macdonald, Black and Aucott69). These same authors published a second meta-analysis which assessed the effect of dietary changes in NAE on urinary Ca, Ca balance, and N-telopeptide, which found that a higher NAE does not reflect a net loss of whole body Ca(Reference Dawson-Hughes, Harris and Palermo70). This analysis had several limitations – the studies included did not directly measure bone health or the progression of osteoporosis, and only studies which modified protein intake were included. Thus, studies which examined the effect of changes in NAE from either bicarbonate salts or altered intakes of fruit and vegetables or grain foods were excluded, and not assessed by this meta-analysis.
Kidney stones
Also of clinical significance is the role metabolic acidosis plays in nephrolithiasis, and its potential connection to bone loss. Urinary Ca excretion is directly proportional to NAE in both stone-formers and normal subjects(Reference Fenton, Eliasziw and Lyon71). In a study of nearly 200 renal stone-formers designed to identify the greatest risk factors for nephrolithiasis, it was the potential acid load of the diet which had the strongest association with stone risk. The authors suggest ‘that a diet with a very low potential acid load should be encouraged in renal stone patients for the prevention of recurrent stones. This result can be obtained by the restriction of animal proteins but also by abundant supplementation with vegetables and fruits’(Reference Trinchieri, Lizzano and Marchesotti28). Potassium-magnesium citrate has been shown to counter renal stone formation associated with immobilisation and was associated with a significant increase in urinary pH(Reference Fenton, Lyon and Eliasziw72). Though increased urinary Mg, pH and citrate are associated with reduced stone formation individually, their effect appears to be greater when used together(Reference Lemann73). Potassium bicarbonate is more effective than potassium chloride for preventing stone formation when used with hydrochlorothiazide(Reference Zerwekh, Odvina and Wuermser74). Potassium citrate is also frequently used, and has been found to reduce bone degradation in stone-formers(Reference Sellmeyer, Schloetter and Sebastian65) and reduce urinary Ca excretion(Reference Sellmeyer, Stone and Sebastian63). The same may be true of potassium and/or magnesium citrate and for stone prevention – in a trial with normal volunteers and stone-formers, potassium and magnesium citrate together had a greater effect than either individually to reduce the ion activity product index of calcium oxalate(Reference Schwille, Schmiedl and Herrmann75). Potassium-magnesium citrate was also shown to reduce the risk of calcium oxalate stone formation by 85 % during a 3-year period in a randomised controlled trial(Reference Frassetto, Nash and Morris76), perhaps by correcting hypocitraturia, a known risk factor for forming kidney stones and a factor (as mentioned previously) inversely associated with acid load calculated by PRAL(Reference Trinchieri, Lizzano and Marchesotti28).
Unlike in the studies of healthy subjects, in both men and women with a history of forming Ca stones, a 2-year treatment with potassium citrate increased forearm bone mineral density in idiopathic Ca stone-formers, with the speculation that it was the alkali load that reduced bone resorption by buffering endogenous acid production(Reference Sellmeyer, Schloetter and Sebastian65).
Muscle
Another area of interest is the use of alkaline therapy for improving muscle function, exercise capacity and reducing age-related muscle wasting. Acidaemia has been shown to increase muscle degradation in patients on haemodialysis(Reference Kato, Yamaguchi and Yachiku77). One epidemiological study of 384 healthy men and women aged 65+ years found a higher intake of foods rich in K (fruits and vegetables) was associated with greater lean muscle mass. The authors speculated that ‘this association is likely to result from the fact that the ingestion of potassium-rich alkaline foods such as fruit and vegetables relieves the mild metabolic acidosis that occurs with the ingestion of a typical American diet’, and suggest that it is plausible that age-related muscle mass decline and sarcopenia may be prevented by the appropriate intake of alkaline K salts(Reference Ettinger, Pak and Citron78).
Potassium bicarbonate has been shown to neutralise the metabolic acidosis, and reduce urinary N wasting in postmenopausal women. As faecal N excretion is typically very low ( < 12 %), and unaffected by alkali administration, the authors speculated that this N-sparing may prevent loss of muscle mass, and may even restore past deficits(Reference Löfberg, Gutierrez and Anderstam79). While it cannot be certain that the decrease in N excretion is directly due to less muscle degradation, it is consistent with previous research suggesting that acidosis stimulates muscle proteolysis by activating proteolytic pathways(Reference Mitch, Medina and Grieber29).
Whether neutralisation of acidosis improves exercise function is unclear, with studies demonstrating both positive and negative results. Short-term intake of sodium bicarbonate helped reduce the exercise-related drop in pH, improved anaerobic performance in a dose-dependent manner(Reference Dawson-Hughes, Harris and Ceglia80), improved intermittent sprint performance(Reference Frassetto, Morris and Sebastian81) and was of ergogenic benefit in the performance of short-term, high-intensity work(Reference Douroudos, atouros and Gourgoulis82). One possible mechanism of action is an increase in plasma pH at rest, providing a delayed onset of intracellular acidification during exercise(Reference Bishop and Claudius83), or by providing additional bicarbonate for an increased buffering capacity, as concluded in a trial involving sodium bicarbonate ingestion by elite swimmers(Reference McNaughton, Backx and Palmer84). Other studies have not demonstrated any effect of short-term bicarbonate supplementation(Reference Raymer, Marsh and Kowalchuk85). Longer-term studies on the effects of alkali neutralisation on muscle function and mass are presently underway (LA Frassetto, personal communication).
Other
There may also be a connection between insulin resistance and acid–base equilibrium, though this relationship is still speculative. Insulin resistance has been associated with a lower urinary citrate excretion, and hypocitraturic patients show greater insulin resistance than normocitraturic Ca stone-formers(Reference Lindh, Peyrebrune and Ingham86). Type 2 diabetes mellitus has been shown to increase the risk of uric acid stone formation, because it causes a lower urinary pH due to impaired kidney ammoniagenesis(Reference Ball, Greenhaff and Maughan87). This lower urinary pH cannot be entirely attributed to a greater BMI or acid intake, though both are factors in determining urinary pH(Reference Cupisti, Meola and D'Alessandro88). Finally, in an evaluation of 148 adults with no kidney stones, participants with the metabolic syndrome had a significantly lower 24 h urine pH than those without, with an incremental reduction in pH associated with the number of metabolic abnormalities present(Reference Daudon, Traxer and Conort89, Reference Cameron, Maalouf and Adams-Huet90).
Also of clinical relevance may be the treatment of pain, although it is less well studied. Local tissue acidosis has been documented in patients with complex regional pain syndrome, and leads to increased pain sensation(Reference Maalouf, Cameron and Moe91). The mechanism for pain sensation may be mediated by acid-sensing ion channels (ASIC), with an increase in ASIC activity in spinal dorsal horn neurons promoting pain by central sensitisation, a mechanism documented in rats(Reference Abate, Chandalia and Cabo-Chan92). ASIC activity may also be induced by NO(Reference Birklein, Weber and Ernst93), and appears to be tightly regulated by pH(Reference Duan, Wu and Yu94). This is an area that lacks significant clinical research.
Conclusion
The lack of consensus for both qualitative and quantitative aspects of acid–base chemistry in physiological systems as well as measurement has caused considerable confusion for both researchers and clinicians. This confusion has also complicated the search for cause and effect and made clinical application difficult and controversial. Nonetheless, the available research makes a compelling case that diet-induced acidosis is a real phenomenon, has significant clinical relevance, may largely be prevented through dietary changes, and should be recognised and treated.
Both dietary interventions (lowering protein and increasing fruit and vegetable consumption) and nutritional supplementation (with K and Mg salts) have been shown to normalise acidosis, but with discordant results on whether this is then associated with clinical improvement in bone, muscle or other physiological or pathophysiological conditions. A positive NEAP diet results in increased urine Ca, N and bone marker excretion, and predisposes to kidney stones. Whether or not, over the longer term, this translates to lower bone density, increased bone and muscle loss with ageing is unclear and requires further investigation.
Acknowledgements
The present review was supported by partial funding from pH Sciences (Seattle, WA, USA).
L. A. F. would like to acknowledge Anthony Sebastian and R. Curtis Morris Jr, the two people she has worked with for all of these years, who are responsible for the ideas behind what she does.
J. P. and J. K. were responsible for data review and writing of the initial manuscript. L. A. F. provided critical review and editing of several drafts, and participated in final manuscript writing.


