



Se is an essential micronutrient for human health( Reference Rayman 1 , Reference Roman, Jitaru and Barbante 2 ). It is incorporated as selenocysteine into approximately twenty-five Se-containing enzymes (selenoproteins), which have been proposed to have fundamental roles in oxidative defence pathways, thyroid hormone production, inflammation and cardiovascular health( Reference Lobanov, Hatfield and Gladyshev 3 ). Se status has also been linked to cancer risk on the basis of cell, animal and epidemiological studies( Reference Hesketh and Méplan 4 – Reference Lipinski 19 ), but clinical studies involving Se supplementation have been inconclusive( Reference Vinceti, Dennert and Crespi 20 ). This reflects our incomplete understanding of the mechanisms by which selenoproteins function in complex interdependent cellular pathways, how these are regulated, the effect of SNP in Se-sensitive genes, which may alter how an individual responds to fluctuations in Se status, and the differences in experimental design and approaches. Inadequate Se and the potential cellular dysfunction associated with it may increase the risk for disease( Reference Méplan and Hesketh 5 , Reference Méplan 7 ). Severe Se deficiency is rare but sub-optimal intake has been suggested to be prevalent in many parts of the world, and the consequences of inadequate Se status in existing diseases and/or the risk for developing disease further is not well understood( Reference Méplan and Hesketh 5 , Reference Méplan 7 ).
MicroRNA (miRNA) are small non-coding RNA, consisting of nineteen to twenty-five nucleotides, which function as regulators of gene expression by affecting translation of mRNA into proteins( Reference Bartel 21 – Reference Hata and Lieberman 25 ). It has been shown that miRNA are critical for the appropriate regulation of fundamental cellular pathways and as such have been implicated in a range of processes that if incorrectly regulated can lead to diseases such as cancer( Reference Roman, Jitaru and Barbante 2 , Reference Méplan and Hesketh 5 , Reference Méplan 7 ). Furthermore, dietary factors are thought to influence miRNA levels and potentially activate abnormal cellular pathways in cancer development( Reference Shah, Kim and Davidson 26 – Reference Shah, Schwartz and Zhao 31 ), and Se status has been shown to affect miRNA levels in the CaCo-2 cell line( Reference Maciel-Dominguez, Swan and Ford 32 ). However, the effect of dysregulated miRNA expression, due to a low-Se environment, on key pathways in cancer development and progression is unclear.
Colon cancer is the third most common cancer in men and the second most common in women globally( Reference Ferlay, Soerjomataram and Dikshit 33 ). The disease is characterised by distinct mutations throughout life in key biological cellular functions such as the Wnt/β-catenin, p53, ERK/MAPK and the DNA mismatch repair pathways( Reference Schneikert and Behrens 34 – Reference Jiricny and Marra 39 ). Several components of these canonical pathways have been linked with abnormal miRNA activity( Reference Schetter and Harris 40 , Reference Hutchison, Cohen and Onyeagucha 41 ) and Se status( Reference Bera, De Rosa and Rachidi 42 – Reference Berry 51 ).
We hypothesised that there are miRNA whose expression is dysregulated in a low-Se environment, and that this dysregulation explains in part the effects of Se status on key biological pathways. To investigate this hypothesis, we used cultured CaCo-2 cells in a low-Se environment for 72 h before supplementation with or without 40 nm Se (as sodium selenite). The concentration of Se used (40 nm) represents a concentration comparable with plasma levels in people with optimal Se status. It has been previously shown that 40 nm is adequate to maximise expression of the Se-sensitive selenoprotein GPX1( Reference Pagmantidis, Bermano and Villette 18 , Reference Bermano, Arthur and Hesketh 52 ). The absolute counts of 800 human miRNA were measured using NanoString technology to characterise the miRNA profile in response to a low-Se environment (<40 nm) compared with a 40-nm Se supplementation. These miRNA data were then assessed in silico, only on the basis of experimentally validated miRNA–mRNA interactions for their effects on biological pathways based on the gene ontology and Reactome databases. The intent of this study was to discover which miRNA were sensitive to low Se so as to provide a robust data set for further functional validation.
Methods
Cell culture and chemicals
Authenticated CaCo-2 human colon adenocarcinoma cells were purchased from the American Type Culture Collection. All cell culture reagents and chemicals were purchased from Life Technologies unless otherwise stated. The cells were cultured before the experiment in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich), supplemented with 1 % non-essential amino acids, 1 % penicillin–streptomycin solution and 10 % fetal bovine serum, and were maintained at 37°C with 5 % CO2 in a humidified atmosphere. A low-Se medium was prepared containing only DMEM, supplemented with 1 % penicillin–streptomycin solution, 5 µg/ml of insulin (Sigma-Aldrich) and 5 µg/ml of transferrin (Sigma-Aldrich).
The effect of Se supplementation in undifferentiated CaCo-2 cells was determined in three biologically independent assays. For each experiment, 5×105 CaCo-2 cells were seeded into three wells of a six-well tissue-culture plate (Corning) and cultured at 37°C with 5 % CO2 in DMEM-STD in a humidified atmosphere for 24 h to facilitate attachment. The medium was then removed from each well and the cells were washed twice with 1 ml of PBS (Life Technologies). The cells were then treated with 2·5 ml per well of fresh low-Se medium. The cells were then cultured at 37°C with 5 % CO2 in a humidified atmosphere for 72 h. The low-Se medium was then removed from each well and the cells were washed with 1 ml of PBS. The cells were then treated with 2·5 ml per well of fresh low-Se medium with or without 7 ng/ml (40 nm) of sodium selenite (Sigma-Aldrich) for 16 h at 37°C with 5 % CO2 in a humidified atmosphere.
Metabolic activity assay
The effect of supplementation of 40 nm Se on the metabolic activity of CaCo-2 cells cultured in low Se for 72 h was measured using the water-soluble tetrazolium cytotoxicity assay (WST-1; Clontech) as described previously( Reference McCann, Rowland and Roy 53 ). Metabolic activity was assessed at T=0 h (low Se for 72 h) and at T=16 h (with or without 40 nm Se, as described above) on three biological replicates (with eight technical replicates each). Absorbance of the formazan dye produced was measured at 450 and 650 nm using a FlexStation 3 Spectrophotometer (Molecular Devices). For all measurements (including the blank), the background absorbance (650 nm) was subtracted from the detection wavelength (450 nm) and these corrected values were used for analysis.
Real-time PCR assay
The expression of the Se-sensitive genes, GPX1 and SEPW1, was quantified using hydrolysis-probe-based real-time PCR. After 16 h of low Se or treatment with 40 nm of sodium selenite, RNA was extracted using the SV Total RNA Isolation System (Promega) according to the manufacturer’s instructions, with the quality and quantity of total RNA determined on the basis of 260:230 and 260:280 absorbance ratios using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). A sample value of 1·8–2·0 for each ratio was considered to be of sufficient quality. The High-Capacity RNA-to-cDNA kit (Life Technologies) was used to prepare 2 µg of complementary DNA (cDNA) from each RNA sample according to the manufacturer’s instructions, and the cDNA samples were stored at –20°C until required.
For real-time PCR analysis, the PrimeTime (Integrated DNA Technologies) assays were used. All PCR (no-template controls and samples) were prepared as quadruplicate 10 µl reactions comprising a 9·0-µl aliquot of master mix (5·0 µl of 2× Kapa Probe Fast Mix (KK4703; Kapa Biosystems), 0·5 µl of 20× Integrated DNA Technologies (IDT) gene assay, 3·5 µl of nuclease-free water (Life Technologies) and 1 µl of cDNA (at a one in ten dilution in nuclease-free water). The thermal profile used was as follows: 95°C for 20 s followed by forty cycles of 95°C for 3 s and 60°C for 30 s. The experiment was completed using a Rotor-Gene 6000 quantitative PCR instrument (Qiagen). These data were normalised to the geometric mean of the β-2 microglobulin (B2M) and the ribosomal protein L19 (RPL19) reference genes, with the application of the comparative quantification method with efficiency correction, and expressed as fold changes (stressed v. unstressed). The efficiencies for all PCR ranged between 1·93 and 2·01, where 2·0 represents 100 % efficiency. qPCR data were ranked and analysed using the Kruskal–Wallis ANOVA method in SigmaStat 12.3 (Systat Software Inc.). Following ANOVA, significantly different means were identified using Tukey’s post hoc test. All data with P<0·05 were considered to be statistically significant.
Profiling of microRNA expression changes
The NanoString nCounter technology( Reference Geiss, Bumgarner and Birditt 54 ) was used to determine the absolute counts of 800 human miRNA (Human v2 miRNA Expression Assay; NanoString Technologies) in 150 ng of RNA from each sample, as described in the real-time qPCR method. The NanoString profiling was completed according to the manufacturer’s instructions and the raw NanoString profiling data were normalised to the geometric mean of the top 100 expressing miRNA and the absolute number of counts for each miRNA was calculated using nSolver version 2.5 analysis software (NanoString Technologies). All data were analysed for statistical significance using Student’s t test in nSolver version 2.5 software. Data having P<0·05 were considered to be statistically significant.
In silico data analyses
To explore the effect(s) of Se-sensitive miRNA on biological pathways, we focused solely on miRNA–mRNA interactions, which were experimentally validated using the DIANA-TarBase version 7.0( Reference Vlachos, Paraskevopoulou and Karagkouni 55 ). These data were then used to assess which pathways they were involved with on the basis of gene ontologies (Gene Ontology database( 56 – Reference Mi, Poudel and Muruganujan 59 )) and reaction events (Reactome database version 57( 60 , Reference Croft, O’Kelly and Wu 61 )). These data were mapped using Ingenuity® Pathway Analysis software (IPA®; Qiagen).
Results
Culturing CaCo-2 cells in low selenium affects cell metabolism and the expression of selenium status markers
To assess the effect of low Se on CaCo-2 viability and to confirm a low-Se environment, we measured metabolic activity (WST-1 assay) and the expression of GPX1 and SEPW1 genes( Reference Pagmantidis, Bermano and Villette 18 , Reference Kipp, Banning and van Schothorst 62 ) (real-time qPCR). The mean metabolic activity of CaCo-2 cells in a low-Se environment was measured to be 79 (sem 6) % of that of CaCo-2 in a 40-nm Se-supplemented environment. The qPCR data show that expression of the GPX1 and SEPW1 genes was significantly lower in a low-Se environment than in the 40-nm Se-supplemented one (–2·93-fold sem 0·84, P=0·010 and –2·73-fold sem 1·02, P=0·013, respectively). As these genes have previously been shown to be highly sensitive to Se supply, these data show that the cells grown in the low-Se medium did have a low-Se status( Reference Pagmantidis, Bermano and Villette 18 ); thus, other Se-sensitive genes may also have responded appropriately to the amount of Se present.
Low selenium affects microRNA expression in CaCo-2 cells
Of the 800 miRNA probes in the NanoString Codeset, the expression of ten miRNA was found to be affected by low Se (Table 1). On the basis of experimentally verified miRNA–mRNA interactions from the DIANA-TarBase version 7.0 database( Reference Vlachos, Paraskevopoulou and Karagkouni 55 ), these ten Se-sensitive miRNA have 5364 interactions with 3588 distinct mRNA.
Table 1 The ten microRNA (miRNA) whose expression is altered by a low-selenium environment in the CaCo-2 cell lineFootnote * (Mean expression levels (EL) with their standard errors)

miRNA–mRNA, the number of unique interactions for a given miRNA within the overall 5364 experimentally supported interactions.
* EL compared with adequate Se (40 nm, EL=1·0), of three independent replicates and was generated using nSolver (version 2.5) software and the DIANA-TarBase version 7.0 database( Reference Vlachos, Paraskevopoulou and Karagkouni 55 ).
Selenium-sensitive microRNA expression affects genes associated with cellular metabolism and signalling
To explore the potential biological implications of the findings from Table 1, we utilised a range of open source databases for our analyses. First, we used the Gene Ontology database( 56 ) to identify the biological processes that the mRNA targeted by Se-sensitive miRNA are involved in, based on Protein ANnotation THrough Evolutionary Relationship (PANTHER) overrepresentation analysis( Reference Mi, Poudel and Muruganujan 59 ) (Fig. 1).

Fig. 1 A heat map showing the gene ontologies affected by expression of selenium-responsive microRNA (miRNA). The intensity of the colours represents the log (P value) of the probability that the association of miRNA–mRNA in a particular pathway occurred by chance. The lower the log (P value) the less likely these associations occur by chance (indicated in red).
These data show that mRNA influenced by Se-sensitive miRNA are involved in key biological processes. While exploring the effects of Se-sensitive miRNA on mRNA on the basis of gene ontology pathways, the findings do not necessarily translate into biological interactions at the protein or the metabolite level. To clarify these effects, we further examined our data using the Reactome database, which uses reactions between entities (e.g. nucleic acids, proteins, complexes and small molecules) as the central point, rather than just gene interactions and associations.
Selenium-sensitive microRNA affect genes associated with the cell cycle and the cellular response to stress
On the basis of Reactome database analyses of the mRNA targeted by the ten Se-sensitive miRNA (Table 1), the cell cycle and the cellular response to stress pathways are, as expected, the most significant pathways affected as a consequence of the dysregulation of Se-sensitive miRNA (Table 2).
Table 2 A summary of the significant high-level reactome pathways containing genes whose expression is influenced by the selenium-sensitive microRNA shown in Table 1 *
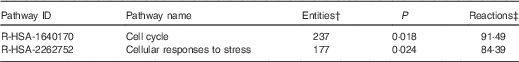
* Data were derived from the Reactome database( 60 ).
† The number of mapped entities, in this case genes that match the pathway.
‡ The percentage of reactions in a specified pathway with at least one mapped entity in the data set.
The genes within the cell cycle and the cellular response to stress pathways, which are experimentally validated targets of the ten Se-sensitive miRNA, are listed in Table 3. These data show that, of the ten Se-sensitive miRNA, mir-93-5p targets the majority of mRNA in these pathways, and that several histone-cluster genes (particularly the histone H4 cluster) are the largest family of mRNA that are targeted.
Table 3 The selenium-sensitive microRNA (miRNA) whose expression is altered by a low-selenium environment and their mRNA targets in the Reactome pathways (Table 2)

FC, fold change.
These data show that considering both gene-based pathway mapping (e.g. gene ontologies) and reaction events (e.g. Reactome data) provides a more comprehensive analyses of the consequences of genes targeted by Se-sensitive miRNA.
Selenium-sensitive microRNA dysregulation may affect appropriate regulation of cancer pathways
To explore the effect of dysregulated Se-sensitive miRNA (Table 1) on genes associated with cancer pathways, we mapped the mRNA targets of these miRNA onto the Wnt/β-catenin, p53 and ERK/MAPK pathways using Ingenuity Pathway Analysis (Fig. 2–4). These data show that several genes, for example, AKT, ERK1/2, MDM2, PIK3, RAS and p53, in key colon cancer-associated pathways are targeted by Se-sensitive miRNA (Table 1), indicating that fluctuations in Se status may affect signalling pathways linked to colon cancer development and progression.

Fig. 2 Effect of dysregulated selenium-sensitive miRNA (Table 1) on genes associated with the Wnt/β-catenin pathway. Genes targeted by selenium-sensitive miRNA are highlighted in yellow. These data were generated through the use of IPA (Ingenuity® Systems, www.ingenuity.com).

Fig. 3 Effect of dysregulated selenium-sensitive miRNA (Table 1) on genes associated with the p53 pathway. Genes targeted by selenium-sensitive miRNA are highlighted in yellow. These data were generated through the use of IPA (Ingenuity® Systems, www.ingenuity.com).

Fig. 4 Effect of dysregulated selenium-sensitive miRNA (Table 1) on genes associated with the ERK/MAPK pathway. Genes targeted by selenium-sensitive miRNA are highlighted in yellow. These data were generated through the use of IPA (Ingenuity® Systems, www.ingenuity.com).
Discussion
In our study, we hypothesised that there are miRNA whose expression is dysregulated in a low-Se environment (<40 nm), and that this dysregulation explains in part the effects of Se status on key biological pathways. We have shown that there are ten miRNA, which have been experimentally validated in the literature, to target mRNA involved in key metabolic and signalling pathways (Table 3) and that canonical signalling pathways in colon cancer are sensitive to Se (Fig. 2–4). To our knowledge, these data are the first reported on Se-sensitive miRNA on the basis of direct miRNA counts rather than on the basis of relative quantification-based technologies such as microarray and real-time qPCR. Furthermore, we explored the pathways/processes that these miRNA targeted, considering only their experimentally validated mRNA targets, on the basis of gene ontologies and Reactome data. Importantly, our data show that the effects of low Se on biological pathways may, in part, be due to the dysregulation of the ten Se-sensitive miRNA identified.
A previous study on the effects of Se status on miRNA used microarray and real-time qPCR methods( Reference Maciel-Dominguez, Swan and Ford 32 ) and found that thirty-four miRNA and fifty mRNA were dysregulated in low-Se conditions in vitro. In contrast to our data, none of the Se-sensitive miRNA reported in Table 1 was found to be affected in that study. However, twenty-eight of these previously observed miRNA were found in our data set, and despite not being differently expressed in our data, the magnitude of the response in expression was consistent. Interestingly, seven of the mRNA (ATP1B3, CACYBP, CMIP, DCUN1D4, GPX4, MT1F and SEPHS2) that showed altered expression are also targets of Se-sensitive miRNA in our data set, despite little overlap in miRNA data( Reference Maciel-Dominguez, Swan and Ford 32 ). Discrepancies between the two studies might be explained by the use of different detection technologies (microarray v. NanoString) and the updated Genome Reference Consortium Human build 38 used in this study.
The data from bioinformatic analysis (Table 3) show that key genes in the cell cycle and the cellular response to stress are regulated by the Se-sensitive miRNA identified in this study. With respect to the cell cycle, genes regulating cell cycle progression, for example, cyclins (B1, B2, E2), cyclin-dependent kinases (1 and 2) and cyclin-dependent kinase inhibitors (1A), checkpoints (E2F1 and RB), and DNA licencing for replication (MCM2 and MCM5) are influenced by Se-sensitive miRNA. Previous studies( Reference Hawkes, Printsev and Alkan 12 , Reference Wu, Cao and Chen 15 , Reference Hawkes and Alkan 16 ) have reported that Se status influences the expression of these genes in disease, and the appropriate regulation of the cell cycle by both mRNA and miRNA is essential for preventing carcinogenic transformation. The bioinformatic analysis also found that Se-sensitive miRNA regulate the expression of genes involved in the cellular response to stress, for example, antioxidant enzymes (SOD2), mitogen-activated signalling cascades (MAPK1, MAPK7 and MAPK8) and in the heat-shock stress responses (HSPA8, HSPA9 and HSPA13). Se status is known to influence several stress responses, such as those caused by oxidative and unfolded protein stress( Reference Wu, Zhang and Dong 63 – Reference Touat-Hamici, Legrain and Bulteau 65 ), and our data suggest that Se-sensitive miRNA may be influential in Se-mediated stress responses.
We have also found that in CaCo-2 cells, inadequate Se supply alters the expression profile of miRNA that regulate the expression of key genes in the canonical Wnt/β-catenin, p53 and ERK/MAPK pathways (Fig. 2–4). Previous studies have shown a link between Se status and the expression of genes in these pathways( Reference Hawkes, Printsev and Alkan 12 – Reference Hawkes and Alkan 16 , Reference Bera, De Rosa and Rachidi 42 , Reference Barrett, Reddy and Short 44 – Reference Xing, Liu and Yang 48 ); however, they did not assess the effect of altered miRNA expression in response to Se status on the genes that were Se sensitive in these pathways. Our data suggest that fluctuations in Se status due to diet or ill health( Reference Méplan and Hesketh 5 , Reference Méplan 7 ), which result in dysregulated miRNA expression, may affect the appropriate functioning of these pathways, influencing pathogenesis in the colon.
Inadequate Se levels and the potential cellular dysfunction associated with them may increase the risk for disease. Se supply can be affected by compromised intestinal function – for example, by inflammatory bowel disease( Reference Kudva, Shay and Prabhu 66 ), or by altered activity of key genes due to SNP( Reference Méplan and Hesketh 5 ) – such that the consequences of fluctuating Se status, exacerbated by inappropriate Se-sensitive miRNA-mediated regulation, may have an important role in how intestinal diseases such as cancer develop and progress.
Although our study has identified that ten miRNA are sensitive to Se status and that these miRNA target mRNA involved in the cell cycle, cell stress response and in key cancer-associated pathways, these findings require further validation. Our data are based on experimental profiling of miRNA abundance and in silico mRNA target analysis (based on experimentally validated miRNA–mRNA interactions, but not necessarily in the cell line used for this study). To robustly demonstrate that these Se miRNA directly influence the mRNA and biological pathways identified in this study, further functional validation, such as protein abundance/activity analysis, is required. However, our data provide a foundation on which to further study the influence of Se status on miRNA-mediated regulation of biological pathways.
Conclusions
Using a novel direct quantification method, we have identified ten miRNA that are Se sensitive, which have been experimentally validated in the literature to target mRNA involved in key metabolic, signalling and canonical colon cancer pathways. Importantly, our data represent important preliminary knowledge of how Se status affects miRNA expression and how these changes may affect biological pathways. First, the data show that the effects of low Se on biological pathways may, in part, be due to the dysregulation of the ten Se-sensitive miRNA identified. Second, the data identify the cell cycle and the stress response pathways as being potentially affected by Se status through miRNA expression. These findings contribute to an understanding of how the regulation of miRNA by Se supply could potentially influence the cells’ response to further challenges that may ultimately lead to disease.
Acknowledgements
This study was co-funded by the MICROGENNET exchange programme funded by the EU Marie Curie International Research Staff Exchange Scheme grant and the Royal Society of New Zealand. Additional funding was provided by AgResearch Ltd (Core funding – Food Nutrition Core programme) and by the New Zealand Ministry of Business, Innovation and Employment (Nutrigenomics New Zealand programme). The funding sponsors had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
M. J. M. and J. E. H. devised the experiments; M. J. M. and K. R. completed the experimental work; M. J. M. completed the data analysis and wrote the manuscript, with direct input from J. E. H., K. R. and N. C. R.
The authors declare that there are no conflicts of interest.








 Open access
Open access