


INTRODUCTION
In humans, typhoid and related diseases commonly called enteric fever, continues to be one of the most serious public health problems worldwide. The World Health Organization estimates that the current annual global burden of typhoid is about 22 million new cases, 5% of which are fatal [Reference Crump, Luby and Mintz1]. Developing countries have a higher incidence of typhoid because of many interrelated factors [Reference Ackers2]. Typhoid fever in endemic areas is common, serious, and increasingly difficult to treat because of resistance to antibiotics [Reference Mermin3].
Different microbiological and serological techniques have been used to diagnose enteric fever. These techniques include culture of blood, bone marrow, faeces, urine [Reference Rockhill4], duodenal string culture [Reference Avendano5], and measurement of specific antibodies by the Widal test [Reference Levine6], ELISA [Reference Carlsson, Lindberg and Hammarsetorm7] and immunofluorescence [Reference Svenungsson, Jorbeck and Lindberg8]. These techniques have limitations in terms of specificity and sensitivity and are being replaced to a great extent by the use of PCR in routine diagnostic laboratories [Reference Emmanuel9–Reference Haque11].
Recently, serotypes of Salmonella other than S. Typhi have been emerging as causes of typhoid in the Indian subcontinent and in the wider world [Reference Hirose12, Reference Baker13]. Moreover, the incidence of enteric fever caused by drug-resistant S. Paratyphi A has markedly increased and in one study represented up to 24% of all isolates [Reference Chandel14]. These reports indicate that PCR assays specific for a single serotype will lack sensitivity for the definitive diagnosis of typhoid in all suspected cases. At the same time Vi-negative variants of S. Typhi have been detected in various parts of the world, especially in Pakistan, with increasing frequency [Reference Baker13]. These variants must have alternative pathogenic mechanisms (as they lack Vi antigen) and thus pose different challenges for treatment. Therefore a molecular diagnostic test giving information about all possible causative agents of typhoidal diseases has the potential to be a very useful tool.
Existing PCR assays for the diagnosis of typhoid fever detect fliC [Reference Song15], Vi [Reference Hashimoto16], groEL [Reference Nair17], or 16S rRNA [Reference Zhu, Lim and Chan18] and are specific for S. Typhi only. To address this problem, a multiplex PCR targeting tyvelose epimerase (tyv) (previously rfbE), fliC-d, paratose synthase (prt) (previously rfbS), viaB and fliC-a (phase-1 flagellin; H:a) genes was developed by Hirose et al. [Reference Hirose12]. The specificity of this assay was reported to be satisfactory but its use is limited to purified bacterial colonies. It lacks the sensitivity to detect directly very small numbers of bacteria that may be circulating in the blood of patients suffering from enteric fever. Its utility is not only limited by the need for culture and isolation of the bacteria, but it exhibited positivity in only 50% of blood culture-positive cases [Reference Guerra19] and this figure is reduced for patients who have already taken an antimicrobial drug.
To overcome these problems and to make this technique directly applicable to patient samples, we designed new internal primers and developed a nested multiplex PCR that proved to be sensitive and specific for the detection of typhoidal pathogens in blood samples.
MATERIALS AND METHODS
Bacterial isolates
Thirty Salmonella isolates from a local population in the Faisalabad region, Pakistan, were selected from the culture collection of the National Institute for Biotechnology and Genetic Engineering (NIBGE). Representative isolates of Escherichia coli, Proteus vulgaris, Pseudomonas aeruginosa, Klebsiella aerogenes, Aerobacter spp., Micrococcus, Staphylococcus, and Streptococcus were also selected as negative controls. Isolates were stored at −20°C in TSB tryptic soy broth (TSB; Merck, Darmstadt, Germany) containing 10% dimethyl sulfoxide, and revived in the same broth as required, and subcultured on MacConkey agar for purity.
Selection of patients
Sixty patients of all ages were included in the study. All cases were admitted to different hospitals in Faisalabad. Diagnostic criteria for typhoid were fever for ⩽3 days with enlarged spleen, headache, malaise, abdominal discomfort, and/or agitation. Patients with severe diarrhoea and/or profuse vomiting were excluded.
Blood culture
Blood samples (2 ml each) were collected in tubes containing anticoagulant (20 mm potassium EDTA) and stored at 4°C and processed for PCR within 48 h of collection. A 2-ml blood sample was inoculated into a culture bottle containing 16 ml TSB with 0·02% SPS (sodium polyanethanol sulfonate) and incubated at 37°C for 72 h. This was subcultured onto MacConkey agar and after incubation overnight, isolated, colourless, smooth colonies of 2–3 mm diameter were selected for further investigation. Species were differentiated by inoculation into triple sugar iron medium (Merck) and results were interpreted according to the manufacturer's guidelines.
Serology
The presence of Vi antigen in S. Typhi isolates was detected by slide agglutination with monovalent Vi antiserum (Bio-Rad, Marnes-la-Coquette, France).
DNA extraction
Total genomic DNA from bacterial isolates was extracted from overnight culture in TSB by the phenol–chloroform method [Reference Sambrook, Fritsch and Maniatis20]. DNA from 200 μl of each blood sample was recovered with the aid of a DNA extraction kit (Fermentas, Hanover, MD, USA) and dissolved in 100 μl distilled water for use as the template in PCR. The integrity and purity of DNA samples were checked by 1% agarose gel electrophoresis and ratio of A260/A280.
Primers
Oligonucleotide primers (Sigma, Dorset, UK) targeting five different genes including tyv, prt, fliC-d, fliC-a [Reference Hirose12] and viaB [Reference Hashimoto16] were used simultaneously for the first round of multiplex PCR (Table 1). The primer sets for nested multiplex PCR (Table 2) were designed from the nucleotide sequence database (GenBank) and PCR conditions were optimized to increase the sensitivity to a level directly applicable on blood samples. The primer sequences and the sizes of their respective amplified products are shown in Tables 1 and 2.
Table 1. Primers for regular multiplex PCR

Table 2. Primers for nested multiplex PCR

Regular multiplex PCR
Each 50 μl of reaction mixture for regular multiplex PCR, in addition to 10 μl of template, contained 1·5 mm MgCl2, 50 nmol of each dNTP, 40 pm of each primer and 5 U of Taq polymerase (Fermentas). The thermal cycler (MasterCycler; Eppendorf, Hamburg, Germany) conditions for 30 cycles were: denaturation at 94°C for 1 min, annealing at 48°C for 1 min and extension at 72°C for 1·5 min.
Nested multiplex PCR
A 1/5 dilution (in sterile distilled water) of amplification product of regular multiplex PCR was used as template. Each 50 μl of reaction mixture for nested multiplex PCR, in addition to 10 μl of template, contained 1·5 mm MgCl2, 70 nmol of each dNTP, 50 pm of each primer and 5 U of Taq polymerase (Fermentas). Cycling conditions for 30 cycles were: denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 1·5 min. The amplification products of both regular and nested multiplex PCRs were separated by electrophoresis in 2% agarose gels, stained, and photographed by a UV transilluminator (Eagle Eye, Stratagene, La Jolla, CA, USA).
Sensitivity of regular and nested PCR
The concentration of DNA was determined spectrophotometrically (μg/μl equal to absorbance at 260 nm multiplied by 10) and extrapolated for the number of bacteria (one S. Typhi bacterium contains 4 fg DNA) according to calculations reported by Song et al. [Reference Song15]. Tenfold dilutions of DNA representing viable counts of bacteria ranging from 107 to 10 c.f.u./ml were made in sterile distilled water and tested by regular PCR. The amplification products were subjected to nested multiplex PCR after 1/5 dilution in distilled water.
PCR for Salmonella
This PCR was used for cross-checking patient blood samples that were positive by multiplex PCR but negative by blood culture. It specifically targets the aroC gene of Salmonella. The primers for regular PCR were 5′-GGCACCAGTATTGGCCTGCT-3′ and 5′-CATATGCGCCACAATGTGTTG-3′ [Reference Kidgell21]. The nested primers were 5′-CCTATGAGCAGAAATACGGC-3′ and 5′-GATTTCATCGCGATTCTGGC-3′. For PCR, each 50 μl of reaction mixture, contained 1·5 mm MgCl2, 40 nmol of each dNTP, 30 pm of each primer and 2 U of Taq polymerase (Fermentas) in addition to template. Cycling conditions for 20 cycles were: denaturation at 94°C for 45 s, annealing at 57°C for 1 min and extension at 72°C for 1·5 min. Conditions were similar for the nested PCR except for annealing temperature (50°C) and extension time (1 min). The size of amplification products was 639 bp and 460 bp for the regular and nested PCR respectively.
RESULTS
Regular and nested multiplex PCR
The regular multiplex PCR showed amplified products of expected sizes (Table 1) for tyv, prt, fliC-d, fliC-a and viaB genes with template DNA from Vi-positive and Vi-negative S. Typhi, and S. Paratyphi A (Fig. 1). In spiking experiments, the limit of detection for this PCR was 105 bacteria/ml.

Fig. 1. Regular multiplex PCR for typhoid associated Salmonella species. Lanes 1 and 2, Vi-negative S. Typhi (fliC-d, tyv and prt genes); lanes 3 and 4, Vi-positive S. Typhi (fliC-d, tyv, viaB and prt genes); lane 5, molecular weight marker; lane 6, negative DNA template control; lanes 7 and 8, S. Paratyphi A (fliC-a and prt genes).
The second round nested multiplex PCR, successfully amplified the same five genes with predicted product sizes (Table 2) from the panel of isolates (Fig. 2). All control non-Salmonella species were negative in the PCR. In the nested PCR of the 30 Salmonella isolates, 21 were Vi-positive S. Typhi (positive for tyv, viaB, fliC-d and prt genes), six Vi-negative S. Typhi and three S. Paratyphi A (prt and fliC-a genes only). The nested PCR detected DNA equivalent to 10 c.f.u./ml (Fig. 3).
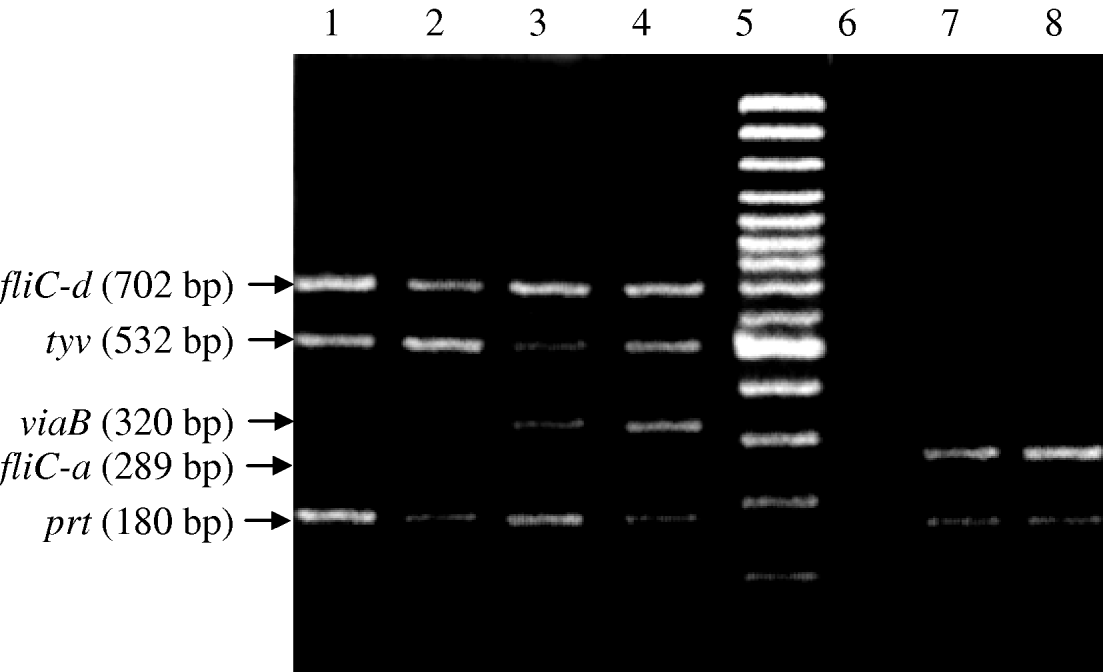
Fig. 2. Nested multiplex PCR for typhoid associated Salmonella species. Lanes 1 and 2, Vi-negative S. Typhi (fliC-d, tyv and prt genes); lanes 3 and 4, Vi-positive S. Typhi (fliC-d, tyv, viaB and prt genes; lane 5, molecular weight marker; lane 6, negative DNA template control; lanes 7 and 8, S. Paratyphi A (fliC-a and prt genes).

Fig. 3. Sensitivity of regular and nested multiplex PCR. Lanes 1–4, regular PCR on DNA of Vi-positive S. Typhi representative of 107, 106, 105 and 104 c.f.u./ml respectively; lane 5, molecular weight marker; lanes 6–10, nested PCR on DNA of Vi-positive S. Typhi representative of 104, 103, 102, 10 and 5 c.f.u./ml respectively.
Diagnosis of suspected typhoid patients
Of the 60 patient cases, 17 were blood culture positive; 16 were S. Typhi and the other was S. Paratyphi A. Four of the 16 S. Typhi isolates were Vi-negative. By nested PCR, 26/60 cases were positive for Vi-positive S. Typhi, nine with Vi-negative S. Typhi and two with S. Paratyphi A. Five formed products consistent with mixed infection of Vi-positive S. Typhi and S. Paratyphi A (Fig. 4). All blood culture-positive cases were detected by PCR. Blood samples from the 25 culture-negative, PCR-positive patients were positive for the aroC gene indicative of Salmonella species [Reference Kidgell21].

Fig. 4. Nested multiplex PCR for Salmonella directly from blood samples. Lanes 1 and 9, S. Paratyphi A (fliC-a and prt genes); lanes 2, 4 and 5, Vi-positive S. Typhi (fliC-d, tyv, viaB and prt genes); lane 6, molecular weight marker; lanes 3 and 10, Vi-negative S. Typhi (fliC-d, tyv and prt genes); lane 7, amplified products from mixed infection by S. Typhi and S. Paratyphi A; lane 8, negative case.
DISCUSSION
Typhoid, usually caused by Vi-positive S. Typhi, is an important health problem especially in the developing countries with about 22 million new cases each year, 5% of which are fatal [Reference Crump, Luby and Mintz1, Reference Hessel22]. However Vi-negative variants of S. Typhi and S. Paratyphi A are emerging as a cause of typhoidal disease. These strains lack Vi antigen which is considered to be the main virulence factor and is the major target for vaccine development [Reference Kossaczka23]. Other pathogenicity mechanisms must therefore play a role in infection and if uncontrolled as a result of poor immune protection these strains may emerge as competitors to Vi-positive S. Typhi. The foremost step towards their control is an early diagnosis.
Conventionally, blood culture and Widal test are the most commonly used techniques for diagnosis. Blood culture ensures the diagnosis in the first week of infection but has poor sensitivity because as few as 10 bacteria/ml can cause disease [Reference Werner24]. The sensitivity of the blood culture is also affected by the host immune response and the intracellular characteristics of S. Typhi [Reference Escamilla, Ugarte and Kilpatrick25]. The Widal antibody test is non-specific and prone to false positivity [Reference Taylor26], and antibodies may take a week or more to reach detectable levels. The Widal test does not distinguish between Vi-positive and Vi-negative S. Typhi.
PCR for the diagnosis of typhoidal diseases is now well established and offers superior sensitivity and specificity compared with conventional methods [Reference Haque11, Reference Song15, Reference Hashimoto16]. However, most PCR protocols detect single pathogens and are inadequate for differential diagnosis. Multiplex PCR has been successfully applied for differential diagnosis of many diseases caused by viruses, bacteria, fungi, and parasites [Reference Rithidech, Dunn and Gordon27]. Hirose et al. [Reference Hirose12] reported a multiplex PCR that could differentiate between typhoidal pathogens. They targeted five genes: prt (present in both S. Typhi and S. Paratyphi A) that encodes CDP-paratose synthase, which converts CDP-4-keto-3,6-dideoxyglucose to CDP-paratose; tyv (present in both S. Typhi and S. Paratyphi A) that encodes CDP-tyvelose epimerase, which converts CDP-paratose to CDP-tyvelose but the described primers target a specific region of the tyv gene of S. Typhi that has been deleted in S. Paratyphi A causing a frameshift mutation that converts codon 4 of tyv to a stop codon [Reference Verma and Reeves28]; viaB which is only found in Vi-positive organisms and is involved in synthesis of the capsule polysaccharide; the fliC-d gene (phase-1 flagellin gene for d antigen [H:d]) which is specific for S. Typhi; and fliC-a gene (phase-1 flagellin; H:a) which is present only in S. Paratyphi A. This PCR proved to be effective for the identification of isolated typhoidal bacteria but it lacked the sensitivity to be directly applicable for blood samples. The need to culture blood samples, isolate the bacteria and perform the PCR incorporates the inherent lower sensitivity of blood culture.
We developed a nested multiplex PCR as an extension of the original PCR by carefully designing highly specific internal primers (Table 2, Fig. 2). PCR conditions were optimized to detect at least 10 c.f.u./ml so that it could be successfully applied to blood samples directly as a diagnostic tool. False-negative results are theoretically highly unlikely owing to simultaneous amplification of prt and tyv genes which are exclusively and universally present in S. Typhi and S. Paratyphi A [Reference Hirose12]. We verified the specificity of our nested PCR procedure by its negative reactions with control species.
The technique was evaluated directly on blood samples of 60 clinically suspected cases of typhoid. PCR detected typhoidal species in 42 cases, only 17 of which were culture positive. The presence of Salmonella in these samples was confirmed with genus-specific primers [Reference Kidgell21]. These results show that the nested PCR has good potential to be a rapid tool for the definitive, differential diagnosis of typhoid and is superior to conventional methods.
ACKNOWLEDGEMENTS
The research facilities for this work were provided by the National Institute for Biotechnology and Genetic Engineering (NIBGE), Faisalabad, Pakistan and the funds were provided by the Higher Education Commission (HEC) of Pakistan.
DECLARATION OF INTEREST
None.






