



Introduction
A high index of suspicion for autoimmune encephalitis motivates testing for neural antibodies, which serve as immensely useful diagnostic biomarkers. Reference Budhram, Dubey and Sechi1 While a positive antibody result can be instrumental in confirming autoimmune encephalitis, negative results may dissuade clinicians from the diagnosis. Comprehensive neural antibody testing for autoimmune encephalitis that incorporates tissue indirect immunofluorescence (TIIF) is therefore generally recommended to maximize sensitivity because more limited testing carries the risk of missing clinically relevant antibodies. Reference Budhram, Dubey and Sechi1,Reference Hahn, Budhram and Alikhani2 We report two patients who were initially suspected of having autoimmune encephalitis but were referred for assessment of mitochondrial disease after limited antibody testing was negative. They were only later definitively diagnosed with autoimmune encephalitis after more comprehensive antibody testing was performed, highlighting the value of comprehensive testing at disease presentation to avoid diagnostic delay.
Patient 1 is a 19-year-old woman who presented with flu-like symptoms followed by subacute onset of encephalopathy with hearing loss, aphasia and focal-to-bilateral tonic-clonic seizures requiring three anti-seizure medications. Brain MRI showed multifocal T2-FLAIR-hyperintense cortical/subcortical lesions (Figure 1A, B). CSF evaluation revealed elevated protein but normal WBC count. Testing for infectious etiologies was unremarkable. Concern for autoimmune encephalitis prompted serum and CSF testing for anti-NMDAR by cell-based assay (CBA), which was negative. Magnetic resonance spectroscopy showed a slight lactate peak in one of the lesions, and she was referred to the Neurometabolic Clinic for possible mitochondrial disease, where she was noted to have cortical deafness and aphasia. Muscle biopsy was unremarkable, so the likelihood of mitochondrial disease was deemed lower, and empiric immunotherapy for possible autoimmune encephalitis was recommended while awaiting mitochondrial gene testing. Intravenous (IV) methylprednisolone and intravenous immunoglobulin (IVIG) were initiated four months after symptom onset and resulted in dramatic clinico-radiographic improvement, although encephalomalacia in areas of previous lesions was noted (Figure 1C, D). She had only brief episodes of language difficulty without loss of awareness particularly when stressed or fatigued, which were possibly of epileptic origin and thought to be related to encephalomalacia. Mitochondrial gene testing was unremarkable, and she was referred to the Epilepsy Clinic for concern of seizures related to autoimmune encephalitis. However, additional serum autoimmune encephalitis antibody testing consisting of anti-NMDAR, LGI1, CASPR2, AMPAR, GABA(B)R and DPPX by CBA was unremarkable, and it was documented that “all antibodies came back negative.” At age 23, she had clinico-radiographic evidence of disease relapse and received IVIG with improvement. Due to persistent concern for autoimmune encephalitis but ongoing diagnostic uncertainty, serum was sent to London Health Sciences Centre (LHSC) for comprehensive neural antibody testing. Mouse TIIF demonstrated staining compatible with anti-GAD65 (Figure 1E, F). Additional testing confirmed high anti-GAD65 levels of 230,128 IU/ml by ELISA as well as weak positivity for anti-GABA(A)R by CBA, both of which have been associated with autoimmune encephalitis and cortical/subcortical lesions. Reference Ren, Ren and Ren3–Reference Gagnon, Savard and Mourabit Amari5 The patient was assessed in the Autoimmune Neurology Clinic at age 25 and diagnosed with neural antibody-associated autoimmune encephalitis. Given the presence of co-existent antibodies potentially suggestive of underlying neoplasm (e.g., thymoma), MRI of the thorax and ultrasound of the abdomen/pelvis were performed and were negative for malignancy. Due to new focal-onset seizures and cortical/subcortical lesions on brain MRI, she received IV methylprednisolone and rituximab, followed by azathioprine as maintenance immunotherapy for relapsing disease. She had no evidence of clinico-radiographic disease activity at six-month follow-up, but she experienced ongoing episodic language difficulties without loss of awareness while on two anti-seizure medications.
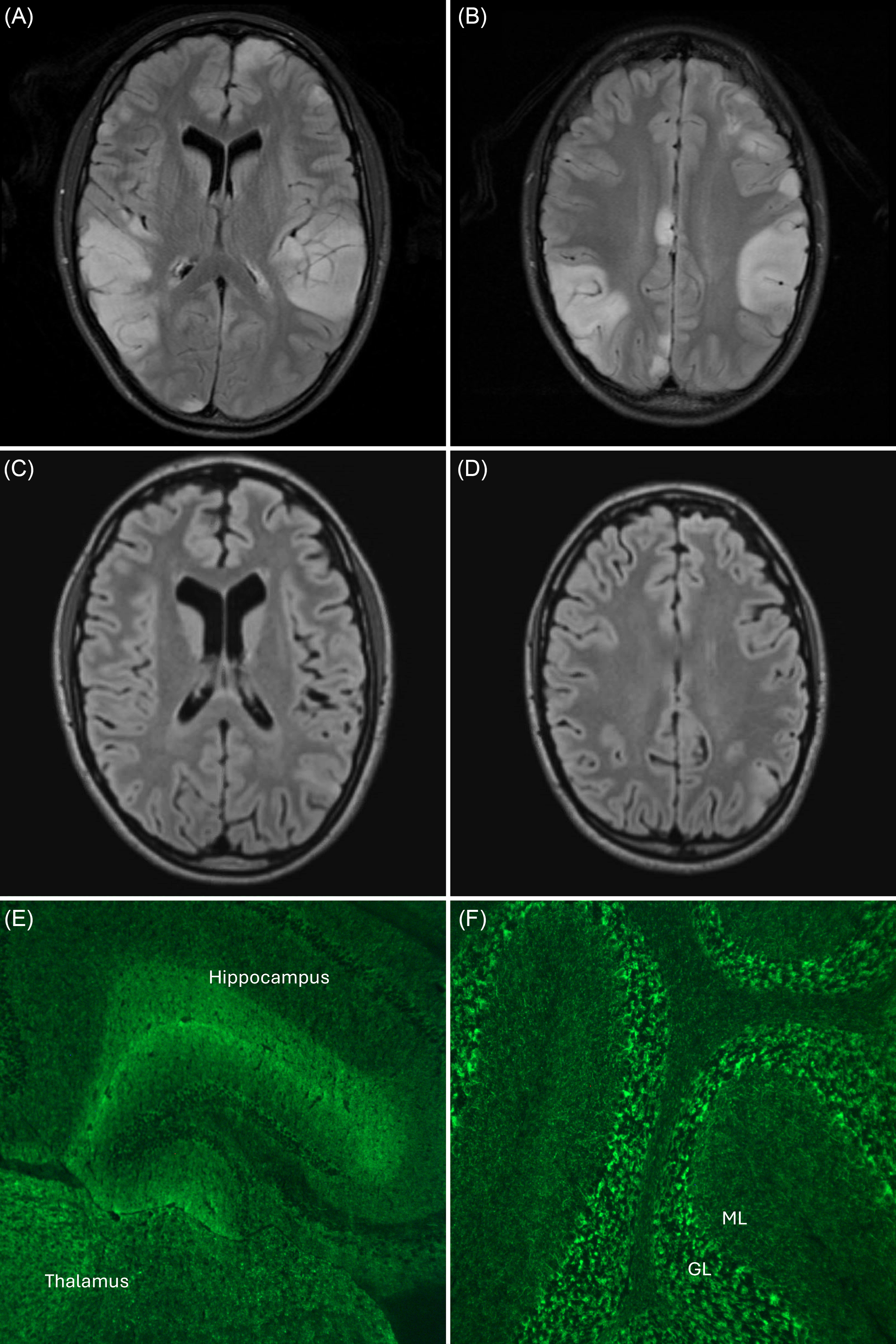
Figure 1. Brain MRI and tissue indirect immunofluorescence of a patient with anti-GAD65 and anti-GABA(A)R-associated autoimmune encephalitis (patient 1). Early in the disease course, multifocal cortical/subcortical T2-FLAIR-hyperintense lesions are seen ( A , B ), without gadolinium enhancement. Repeat brain MRI four months later shows near-resolution of lesions following immunotherapy but with residual encephalomalacia ( C , D ). Mouse tissue indirect immunofluorescence demonstrates staining of the hippocampus, thalamus and cerebellum that is compatible with anti-GAD65 ( E , F ). Additional serum testing confirmed high anti-GAD65 levels of 230,128 IU/ml by ELISA as well as weak positivity for anti-GABA(A)R by cell-based assay. GL = granular layer of the cerebellum; ML = molecular layer of the cerebellum.
Patient 2 is a 26-year-old man who developed a rapidly progressive cerebellar syndrome with encephalopathy. Brain MRI was unremarkable, without cerebellar T2-FLAIR hyperintensity or atrophy (Figure 2A, B). CSF evaluation revealed lymphocytic pleocytosis and elevated protein. Testing for infectious etiologies was unremarkable. Serum paraneoplastic antibody testing consisting of anti-Hu, Yo, Ri, amphiphysin, CV2, Ma2/Ta, Tr, GAD65, SOX1, Zic4, recoverin and titin by immunoblot was unremarkable, and it was documented that “reassuringly all of his antibodies turned out to be negative.” He had a mildly elevated blood lactate level and was referred to the Neurometabolic Clinic for possible mitochondrial disease, where he was noted to have a pan-cerebellar syndrome with ataxic dysarthria, limb and gait ataxia requiring a wheelchair. Repeat brain MRI showed interval development of cerebellar atrophy (Figure 2C, D). The likelihood of mitochondrial disease was deemed low, and empiric immunotherapy with oral prednisone for possible autoimmune encephalitis was initiated five months after symptom onset. Due to concern for autoimmune encephalitis but ongoing diagnostic uncertainty, serum was sent to LHSC for comprehensive neural antibody testing. Mouse TIIF demonstrated staining compatible with anti-mGluR1 (Figure 2E, F), and additional testing confirmed positivity for anti-mGluR1 by CBA, which has been associated with autoimmune cerebellar ataxia. Reference Lopez-Chiriboga, Komorowski and Kümpfel6 The patient was assessed in the Autoimmune Neurology Clinic and diagnosed with neural antibody-associated autoimmune encephalitis. Body CT and testicular ultrasound followed by body positron emission tomography (PET) were negative for malignancy. He received IV methylprednisolone and IVIG followed by rituximab due to ongoing severe ataxia, with mild improvement in cerebellar symptoms at one-month follow-up.

Figure 2. Brain MRI and tissue indirect immunofluorescence of a patient with anti-mGluR1-associated autoimmune encephalitis (patient 2). Early in the disease course brain MRI is unremarkable, with no cerebellar T2-FLAIR hyperintensity or atrophy seen ( A , B ). Repeat brain MRI five months later shows interval development of cerebellar atrophy ( C , D ). Mouse tissue indirect immunofluorescence demonstrates staining of the hippocampus, thalamus and cerebellum that is compatible with anti-mGluR1 ( E , F ). Additional serum testing confirmed positivity for anti-mGluR1 by cell-based assay. GL = granular layer of the cerebellum; ML = molecular layer of the cerebellum.
We describe two patients with autoimmune encephalitis who initially tested negative on more limited antibody testing, which contributed to diagnostic delay. Both patients developed visible sequelae of neuroinflammation on neuroimaging, highlighting the importance of prompt diagnosis and treatment to mitigate irreversible structural injury. Negative antibody results may significantly reduce suspicion for autoimmune encephalitis but, as illustrated by our cases, can be falsely reassuring if only limited testing has been performed. For this reason, when encountering “negative” antibody results in a patient with a high index of suspicion for autoimmune encephalitis, clinicians should critically examine test comprehensiveness. Dozens of antibodies have now been associated with autoimmune encephalitis and more comprehensive testing with the incorporation of TIIF aids in their detection. Reference Budhram and Flanagan7,Reference Gilligan, McGuigan and McKeon8 Importantly, certain antibodies that associate with discrete encephalitic presentations but do not reliably stain tissue (e.g., anti-MOG, GlyR) are often not included in autoimmune encephalitis panels, and certain test methodologies that can further improve sensitivity (e.g., live CBA) may only be available at specialized research laboratories; for this reason, if in doubt about test comprehensiveness in a patient with a high index of suspicion for autoimmune encephalitis, clinicians are encouraged to contact their testing laboratory to discuss. Reference Hahn, Budhram and Alikhani2,Reference Budhram and Flanagan7 It bears emphasizing that antibody-negative autoimmune encephalitis may also occur and benefit from prompt immunotherapy, although comprehensive neural antibody testing, as well as application of proposed syndrome-based criteria, is recommended prior to assigning this diagnosis. Reference Dalmau and Graus9 Our cases highlight the value of comprehensive neural antibody testing for suspected autoimmune encephalitis to reduce the risk of missed antibodies contributing to diagnostic delay.
Author contributions
JA: conceptualization, methodology, investigation, writing – original draft. MAT: conceptualization, writing – review and editing. JGB: conceptualization, writing – review and editing. AB: conceptualization, methodology, investigation, writing – original draft, supervision.
Funding statement
None.
The authors have not published, posted, or submitted any related manuscripts from the same study.
Anonymized data generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request by any qualified investigator.
Competing interests
Dr Al-Tawari has no disclosures to report.
Dr Tarnopolsky reports that he holds an endowed Chair in Neuromuscular and Neurometabolic Disorders from the Hamilton Health Sciences Foundation.
Dr Burneo reports that he holds the Jack Cowin Endowed Chair in Epilepsy Research at Western University.
Dr Budhram reports that he holds the London Health Sciences Centre and London Health Sciences Foundation Chair in Neural Antibody Testing for Neuro-Inflammatory Diseases and receives support from the Opportunities Fund of the Academic Health Sciences Centre Alternative Funding Plan of the Academic Medical Organization of Southwestern Ontario).



 Open access
Open access