


Phenylketonuria is a recessively inherited metabolic disorder which, unless it is treated early enough with a phenylalanine-restricted diet, leads to severe intellectual disabilities. Reference Koch and de la Cruz1 The overall prevalence of phenylketonuria in the UK is about 1 per 10 000 Reference Woolff, Smith, Carson, Graham, Kromrower, Turnbull, Clayton, Mahler and Sutherland2 and published guidelines suggest that treatment needs to be early and lifelong. Reference Waisbren, Schnell and Levy3
Neonatal newborn screening for phenylketonuria began in the late 1960s and those treated early had a very good outcome. Reference Koch and de la Cruz1 However, those born before neonatal screening began were not normally treated, as they already had severe intellectual disabilities, assumed to be irreversible. Our study aimed to trace all those with untreated phenylketonuria and severe intellectual disabilities in the UK and to examine their range of difficulties, as a prelude to a randomised controlled trial of phenylalanine-restricted diet in people with previously untreated phenylketonuria.
Methods
A letter and response form were sent to all UK professionals in a number of fields (psychiatrists, psychologists and managers of learning disability services; dietitians in mental health/learning disabilities services and metabolic services; metabolic paediatricians) asking whether they knew of anyone with untreated phenylketonuria and intellectual disabilities. The survey was also publicised through the National Society for Phenylketonuria website and at conferences. The study was approved by a multisite National Health Service ethics committee.
Professionals were asked for an anonymised list of people known to them with untreated phenylketonuria, plus a contact name of someone who could give further details. These ‘primary contacts’ were then asked to complete a brief anonymised questionnaire (described below) regarding the person with untreated phenylketonuria. To ensure the maximum response rate two reminder letters were sent to primary contacts if there was no reply. In 11% of cases, two contacts completed the questionnaire regarding the same person to provide a measure of interrater reliability (72%).
The questionnaire included questions on: age and gender of the person with untreated phenylketonuria; their relationship to the respondent; and whether the person with phenylketonuria had ever tried a low-phenylalanine diet – if so, for how long. Details of levels of skills, support required, challenging behaviour, and other symptoms were sought. (Copies of the questionnaire can be obtained from G.H.M.)
Most data were nominal or ordinal. Analysis was by non-parametric statistics, including chi-square. Reference Howell4
Results
Of over 500 letters sent out, 194 replies were received. Of these, 77 were positive replies from professionals who knew of one or more adults with untreated phenylketonuria; 117 said they knew of no one with untreated phenylketonuria. Questionnaires were then sent to the primary contacts named in the positive replies and 98 completed questionnaires were returned (84% response rate), regarding adults who had all been untreated in early life.
Of these 98 people with phenylketonuria, none of whom had been treated in infancy, 50 had never tried the phenylalanine-restricted diet at all. The remaining 48 had tried the diet at some point in their lives: 29 had ceased the diet but 19 were still on it.
Of the 79 untreated people (i.e. 50 never on diet + 29 tried it but not on diet now), 44% were men. The overall mean age was 47.7 years (s.d.=9.6, range 19–72). The two youngest people were sisters, aged 19 and 22 years, who had come to the UK from a country that did not have neonatal screening for phenylketonuria; the next youngest person was 34 years old. Of the 29 people who had tried the phenylalanine-restricted diet at some point in their lives, about 50% had tried it for less than 2 years.
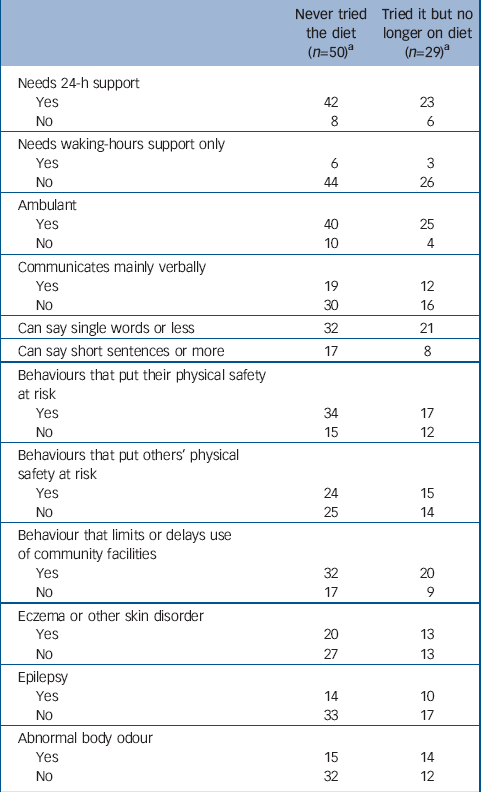
The characteristics of the 79 untreated people and their challenging behaviour are shown in Table 1 (cross-tabulated against whether or not they had ever tried the phenylalanine-restricted diet). There were no significant differences between those who had and who had not tried the diet in terms of their characteristics, challenging behaviour or symptoms.
Table 1 Characteristics, challenging behaviour and symptoms of people with untreated phenylketonuria, not on low-phenylalanine diet
| Never tried the diet (n=50)a | Tried it but no longer on diet (n=29)a | |
|---|---|---|
| Needs 24-h support | ||
| Yes | 42 | 23 |
| No | 8 | 6 |
| Needs waking-hours support only | ||
| Yes | 6 | 3 |
| No | 44 | 26 |
| Ambulant | ||
| Yes | 40 | 25 |
| No | 10 | 4 |
| Communicates mainly verbally | ||
| Yes | 19 | 12 |
| No | 30 | 16 |
| Can say single words or less | 32 | 21 |
| Can say short sentences or more | 17 | 8 |
| Behaviours that put their physical safety at risk | ||
| Yes | 34 | 17 |
| No | 15 | 12 |
| Behaviours that put others' physical safety at risk | ||
| Yes | 24 | 15 |
| No | 25 | 14 |
| Behaviour that limits or delays use of community facilities | ||
| Yes | 32 | 20 |
| No | 17 | 9 |
| Eczema or other skin disorder | ||
| Yes | 20 | 13 |
| No | 27 | 13 |
| Epilepsy | ||
| Yes | 14 | 10 |
| No | 33 | 17 |
| Abnormal body odour | ||
| Yes | 15 | 14 |
| No | 32 | 12 |
The data were also examined for the 19 people, untreated in infancy, who later tried the phenylalanine-restricted diet and were still on the diet. This group did not differ significantly from the 79 people who were not on the diet in terms of their age, gender, levels of skills, challenging behaviours or symptoms.
Discussion
It has been calculated from the general birth rate (prior to neonatal screening for phenylketonuria), together with the known incidence rate for phenylketonuria, that there are about 2000 people with untreated phenylketonuria still alive in the UK, assuming a life expectancy of 65 years. Reference Brown and Guest5 Jancar Reference Jancar6 argued that actual life expectancy for this group is rather lower at 57 years, but even if this were the case, we would expect about 1500 people with untreated phenylketonuria. In fact, fewer than 150 people were found. Although it is possible that some people with mild phenylketonuria are living relatively normal lives and are not in contact with services, it is likely that many people with untreated phenylketonuria and severe intellectual disabilities live in the community, known to learning disability services, but are not recognised as having untreated phenylketonuria. An alternative possibility is that our survey strategy missed very large numbers of people with untreated phenylketonuria (for example, through non-response of professionals), but it is difficult to see what survey method might have been better.
This survey of a relatively large number of people with untreated phenylketonuria clearly documents the extent of their disabilities and the significant support needs they have. Their identification is important as specific intervention with a phenylalanine-restricted diet may potentially reduce these burdens, even at this late stage, as a number of single-case and small-scale studies have suggested. Reference Hoskin, Sasitharan and Howard7–Reference Koch, Moseley, Ning, Romstad, Guldberg and Guttler10 In our study, there were no significant differences in the levels of disabilities, challenging behaviours and symptoms between those who had tried the diet at some point in their lives (whether or not they were still on diet) and those who had never tried it. However, neither ours nor previous studies were prospective randomised controlled trials with standardised measures.
There are two main implications of this study. First, general practitioners, psychiatrists, neurologists, psychologists, community nurses and dietitians, who may see people with severe intellectual disabilities of unknown cause, should suspect and screen for phenylketonuria, especially if the person is more than 35 years old (or was born in a country without neonatal screening), needs 24-h support, and has challenging behaviours and symptoms of phenylketonuria (eczema, epilepsy, ‘mousey’ body odour, and a fair complexion). The second implication is that there is a need for a properly designed randomised control trial to examine whether a phenylalanine-restricted diet really is of help to this very disabled group of people.
Acknowledgements
We are grateful to all at SHS International, especially Pat Portnoi, for their help with this survey, and to the Wellcome Trust for funding.


 Open access
Open access
eLetters
No eLetters have been published for this article.