



Introduction
Globally, tuberculosis (TB) is the leading cause of death among infectious diseases, given that 10 million people became ill in 2017. An estimated 1.6 million people died because of the disease (including 300 000 among people with HIV). Drug-resistant TB (DR-TB) is a continuing threat with 558 000 new cases with resistance to rifampicin (RIF), the most effective first-line drug, of which 82% had multidrug-resistant TB (MDR-TB), and only 160 684 DR-TB were detected. Among the estimated cases of DR-TB, approximately 60% occur in the BRICS countries (Brazil, Russia, India, China and South Africa) [1].
The management of MDR-TB is characterised by delayed diagnosis, uncertainty of the extent of bacillary drug resistance, imprecise standardised drug regimens and dosages, very long duration of therapy and high frequency of adverse events associated with a high morbidity and mortality [2]. Recently, in a meta-analysis of 74 studies, including 17 494 DR-TB participants [Reference Bastos, Lan and Menzies3], the pooled treatment success rate was 26% in extensively drug-resistant TB (XDR-TB) patients and 60% in MDR-TB patients. MDR-TB is characterised as a resistance to isoniazid (INH) and RIF, and XDR-TB is defined as a resistance to all first-line drugs plus at least one fluoroquinolones and second-line injectable (kanamycin, amikacin or capreomycin) [1].
The rpoB (RNA polymerase gene) accounts for 96% of RIF resistance. RIF monoresistance is rare and occurs mostly together with resistance to other drugs, usually INH, making RIF a good marker for MDR [Reference Dookie4]. The most common INH resistant mutation is in katG (catalase peroxidase gene), which accounts for 64.2% and inhA (enoyl-acyl reductase gene) with 19.2% of INH resistance [Reference Gegia5].
Different molecular tools have been developed for Mycobacterium tuberculosis (M.tb) genotyping. Spoligotyping and Mycobacterial interspersed repetitive units of a variable number of tandem repeats (MIRU-VNTR) applied together have been used, generating satisfactory discriminatory power for strain lineage classification [Reference Allix-Béguec6]. More recently, greater access to the drug sensitivity testing (DST) has been proposed through gene sequencing platforms aimed at clinical decision making (e.g. replacing phenotypic DST) [Reference Papaventsis7]. These tools allow an epidemiological investigation of TB transmission and relationship between strains lineage [Reference Chatterjee8].
Strain variation in M.tb complex can have different phenotypic consequences, such as difference in gene expression, growth rates and metabolic responses [Reference Coscolla and Gagneux9]. The genetic background of M.tb strain can also affect the outcome of the infection and the response to drug resistance [Reference Gagneux10]. A molecular understanding of MDR strains through the analysis of resistance-conferring mutations and identification of lineages of M.tb in each region, as well as its interaction with different scenarios, enables a better comprehension of the dynamics of the infection, thus helping the improvement of the TB surveillance worldwide, also contributing with data for new treatment approaches [Reference Kato-Maeda, Metcalfe and Flores11].
Rio Grande do Sul (RS), in the southern region of Brazil, is a high-burden state, currently in fourth place in TB incidence rate among the Brazilians states with 39.5/100 000 cases and MDR-TB cure rate of 51.5% [12]. Among Brazilian states, primary and acquired MDR-TB rates were highest in RS, at 2.2% and 12%, respectively [Reference Micheletti, Kritski and Braga13]. Therefore, the objective of this study was to characterise MDR M.tb isolates from RS state through the analysis of katG, inhA and rpoB genes, as well as RDRio sublineage identification and MIRU-VNTR 24loci and spoligotyping methods.
Methods
Study population and M.tb culture
The M.tb isolates were collected from sputum samples of patients treated at the Hospital Sanatório Paternon (HSP) (a reference centre for TB resistance treatment of the State), during the years of 2013 and 2014. In this period, 18-year-old or older eligible participants, presenting coughs for 3 weeks or more and those with DST performed and presenting bacteriological confirmation of TB, and at least, one of the following conditions for defining them as presumed DR/MDR-TB: (a) with previous anti-TB treatment: being suspected of re-treatment failure or treatment defaults; (b) without previous anti-TB treatment: being HIV-seropositive subjects, or close contact with smear-positive MDR-TB cases. Subjects were excluded if they presented: (a) confirmed drug-sensitive TB, (b) refused to sign the Informed Consent (c) or harboured atypical mycobacteria.
During 2013 and 2014, HSP notified, in the SITE-TB website (database of DR-TB cases reported in Brazil), 186 MDR-TB cases. These 186 drug-resistant samples were submitted to the DST using the liquid MGIT-960 SIRE kit (Becton Dickinson Diagnostic Systems, Sparks, MD, USA) to first-line anti-TB drugs; RIF (1.0 µg/ml), INH (0.1 µg/ml), ethambutol (EMB) (5.0 µg/ml) and streptomycin (STR) (1.0 µg/ml) at the State Central Laboratory (LACEN), the TB diagnosis reference laboratory of the RS State. In this study, 131 MDR M.tb isolates were included. The project was approved by the Research Ethics Committee of the Fundação Estadual de Produção e Pesquisa em Saúde (FEPPS/RS), protocol number 1.587.621 CAAE: 18269313.0.0000.5320.
DNA extraction
The genomic DNA of M.tb was extracted from sputum culture in Lowenstein–Jensen solid medium using Cetyltrimethylammonium Bromide (CTAB) method, as described by van Embden et al. [Reference Van Embden14].
Spoligotyping
Spoligotyping was performed using the Beamedex microsphere technique (Beamedex SAS, Orsay, France) in the Luminex-Bioplex-BioRad 200 system (Luminex Corporation, Austin, TX, USA), developed in the ‘Institut de Genétique et Microbiologi e Université Paris-Sud’, following the protocol described by Zhang et al. [Reference Zhang15]. The definition of the family, lineage level and definition of the spoligotype international type were performed by comparison with profiles deposited in the SITVITWEB database (http://www.pasteur-guadeloupe.fr:8081/SITVIT_ONLINE); when we found unknown or orphan profiles in SITVITWEB, we used SpotClust database to classify the isolates (http://tbinsight.cs.rpi.edu/run_spotclust.html).
MIRU-VNTR 24loci
MIRU-VNTR 24loci was performed as described by de Beer et al. [Reference de Beer16]. Fragment size of the amplicons was analysed on an ABI 3130xl DNA sequence analyser (Applied Biosystems, Foster City, California, USA) and the number of copies of each locus was determined by automated assignment using the Genemapper 3.2.1 software (Applied Biosystems). Undefined results or locus that did not amplify were double checked on agarose gels comparing to a reference table described by Supply et al. [Reference Supply17].
Lineage identification of M.tb isolates was carried out by best match analysis and Tree-based identification tools on the MIRU-VNTRplus database (https://www.miru-vntrplus.org) [Reference Allix-Béguec6]. We applied 0.3 maximum tolerance of difference of four loci, as recommended by the MIRU-VNTRplus website for secure classification [Reference Allix-Béguec6].
The discriminatory power was determined by the Hunter–Gaston discriminatory index (HGDI) [Reference Hunter and Gaston18] calculated online by using the discriminatory power calculator tool available at http://insilico.ehu.es. The allelic diversity of each of the MIRU-VNTR 24loci was determined at the MIRU VNTRplus, classified into highly (HGDI > 0.6), moderately (HGDI > 0.3) or poorly discriminative (HGDI < 0.3). The recent transmission rate was estimated by the N − 1 method [Reference Glynn, Vynnycky and Fine19], according to the formula: number of clustered isolates − number of clusters/total number of isolates.
The classification of the isolates by MIRU-VNTR and spoligotyping was performed based on dendrogram provided in the MIRU-VNTRplus by the construction of a Neighbour-Joining based phylogenetic tree [Reference Allix-Béguec6].
katG, inhA and rpoB sequencing
Sequencing of hotspot regions of genes katG, inhA and rpoB was performed according to Dalla Costa et al. [Reference Dalla Costa20]. Mutation analysis was conducted using SeqScape® and Chromas® software, and also the online platform Blast (http://blast.ncbi.nlm.nih.gov).
RDRio
RDRio sub-lineage was performed using the protocol described by Lazzarini et al. [Reference Lazzarini21] and analysed on 1.5% agarose gel.
Results
Patients' characteristics
Among the 131 MDR-TB cases included, 100 (76.3%) were male, 72 (55%) were in an age bracket of 26–45 years, 124 (94.7%) with pulmonary TB, 57 (43.5%) were current tobacco smokers, 54 (41.2%) were illicit drug users, 37 (28.2%) had alcoholism and 33 (25.1%) had AIDS. Ninety-one (69.5%) had TB in the past; among them, 35 (26.7%) with a history of failure (Table 1).
Table 1. Socio-demographic characteristics of 131 MDR TB patients
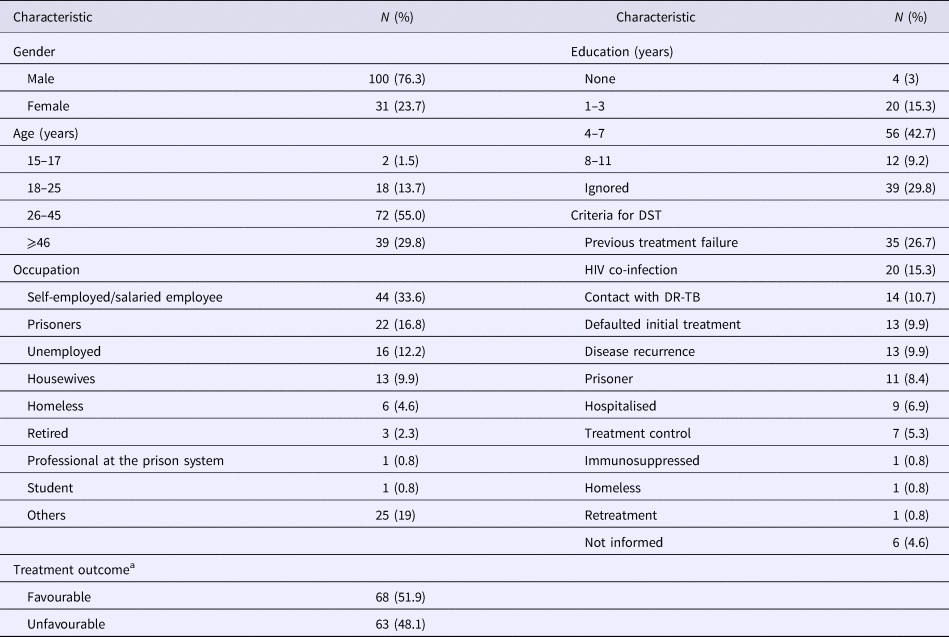
MDR-TB, multidrug-resistant tuberculosis. DR-TB, drug-resistant tuberculosis.
a Favourable treatment outcome: cured patients and patients that completed anti-TB treatment. Unfavourable treatment: patients that abandoned or failure to anti-TB treatment, died by TB or other cause.
Seventeen patients (13%) were additionally resistant to STR, 13 (10%) resistant to EMB and four (3%) resistant to both STR and EMB. The criteria for submission to DST are described in Table 1. Anti-TB treatment outcome, favourable and unfavourable results were identified in 68 (51.9%) and 63 (48.1%), respectively (Table 1).
Mutations
Even though all 131 M.tb isolates were considered MDR according to the DST, eight (6.1%) did not present mutations in the katG, inhA or rpoB genes. In 116 (88.5%) M.tb isolates, we found mutations associated with RIF resistance (in the 511, 512, 513, 516, 526, 531 codons in the rpoB gene), and in 112 isolates (85.5%), we observed mutations related to INH resistance (in the 315 codon in the katG gene and −15 nucleotide of the inhA promoter region) (Table 2). We also found an insertion of 12 nucleotides (CCAGAACAACCC) at the 516 codon in the rpoB gene in 19 (14.5%) isolates. The mutation −17 (T-G) in the inhA gene was not found.
Table 2. Genotypic profile of 131 M.tb isolates

WT, wild type.
Codons change and legend: KatG S315T = AGC → ACC, katG S315R = AGC → AGA, katG S315T = AGC → ACA, katG S315N = AGC → AAC, rpoB L511P CTG-CCG, rpoB S512T AGC-ACC, rpoB Q513E CAA-GAA, rpoB D516Y = GAC → TAC, rpoB D516V = GAC → GTC, rpoB H526Y = CAC → TAC, rpoB H526D = CAC → GAC, rpoB H526C = CAC → TGC, rpoB H526N = CAC → AAC, rpoB S531L = TCG → TTG, rpoB S531W = TCG → TGG, rpoB S531F-L = TCG → TTC-TTG (double peak).
Spoligotyping
With a clustering rate of 0.61 and a discriminatory power of HGDI of 0.68, the 131 M.tb isolates analysed by spoligotyping were classified as the following: 66 (50%) LAM family (LAM 1, LAM2, LAM3, LAM4, LAM5, LAM6, LAM7, LAM9), 23 (17.5%) T family (T1, T2, T5-Madrid), 20 (15.2%) H family (H1, H3), 13 (9.9%) PINI2 family, two (1.5%) X family (X2), two (1.5%) Family33, two (1.5%) Family34, one (0.7%) Family36, one (0.7%) Atypic family and one (0.7%) was classified as S family.
MIRU-VNTR 24loci
In the analysis by MIRU-VNTR, the clustering rate was 0.13 and the discriminatory power of HGDI was 0.66. Out of the 131 isolates, we found 64 of them (48.8%) classified as LAM family, 24 (18.3%) Haarlem family, two (1.5%) X family, two (1.5%) H37RV family, one isolate each (0.8%) were Uganda family, S family, Cameroon family and Delhi/CAS family. It was not possible to classify the families of 35 of these isolates (26.7%) within the 0.3 relaxed value recommended by the MIRU-VNTRplus in the identification by similarity search, but it was possible through the tree-based identification in combination with the spoligotyping patterns (Supplementary S1).
Out of the 24 loci, the highest allelic diversity indices (h > 0.6) were for QUB-26, QUB-11b, Mtub04, MIRU 23, MIRU 40, MIRU 26 and Mtub21. The locis MIRU 02, MIRU 04, Mtub29, MIRU 39 and MIRU 24 presented low allelic diversity indices (h < 0.3) (Supplementary S2).
MIRU-VNTR and spoligotyping combined analysis
A combined analysis of MIRU-VNTR and spoligotyping through the MIRU-VNTRplus was also conducted to better classify the isolates, the distribution of the lineage was: 86/131 (65.6%) belonging to the LAM family, 29/131 (22.1%) Haarlem family, 5/13 (3.8%) Uganda family, 4/131 (3%) were X family, 4/131 (3%) H37RV family and with 1/131 each (0.8%) belonged to the S family, TUR family and West African (Supplementary S1). The discriminatory power of HGDI was 0.52 and the recent transmission index related to the clustering rate was 6.8%.
RDRio
The deletion RDRio was present in 32% of our isolates (42/131), while 67.9% were WT (89/131). Among the isolates that presented the deletion, 40 of them were classified as LAM family, one was TUR and another one Uganda in the combined analysis of MIRU-VNTR and spoligotyping. The LAM sublineage classified through spoligotyping were LAM 5 (14 isolates), LAM 9 (nine isolates), LAM 1 (six isolates), LAM 4 (six isolates) and LAM 2 (three isolates).
We investigated the possible correlation between treatment outcome with the different M.tb lineages and treatment outcome with different profiles of katG, inhA and rpoB mutations, using the logistic regression model. To analyse the possible association between the RDRio genotype and treatment outcome, we used the χ 2 test. Considering the P-value < 0.05, no correlation was found in both statistical analyses.
Discussion
RS is a high-burden state currently in fourth place in TB incidence rate among the Brazilians states, and the city of Porto Alegre is the capital with more TB retreatment cases registered (31.2% of all cases) in 2017 [12]. For a better understanding of the distribution of the MDR strains and the frequency of mutation-associated resistance in the State, we performed genotyping through MIRU-VNTR 24loci and spoligotyping of 131 MDR M.tb isolates, as well as sequencing of drug resistance mutations.
By sequencing the katG gene, mutations in 106 of our isolates (80.9%) at 315 codons were observed, agreeing also with other studies from the same state [Reference Esteves22] and other regions [Reference Dookie4]. Regarding the inhA gene, 41 MDR M.tb isolates (31.3%) had mutation in the promoter region of the gene. The frequency of mutations related to the inhA gene varies from different places [Reference Seifert23], even when compared to sampling from the same region [Reference Esteves22]. In this study, we did not find mutation on −17 position, as described recently in the same state [Reference Esteves22].
Out of the 26 isolates (19.8%) that did not show any mutation in the katG gene, seven of them had mutation in the inhA promoter region, the other 19 (14.5%) presented no mutations in both genes. Furthermore, 90 isolates (68.7%) showed no mutation in the inhA gene, indicating a low mutation rate in this gene in accordance with Zhang and Yew [Reference Zhang and Yew24]. Some studies associated the high frequency of katG mutations with MDR isolates and inhA mutations with monoresistant isolates [Reference Hazbón25]. A possible explanation for this would be that mutations in the promoter region of the inhA region generate a greater biological cost to the bacillus, and the isolates with the mutation in the katG gene would be selected to survive. Thus, the presence of mutation in katG provides to the bacillus greater probability of M.tb isolate to evolve to MDR [Reference Hazbón25, Reference Monteserin26]. Our findings corroborate this hypothesis, since we also found a high frequency of katG mutations and low frequency of inhA mutations among our MDR M.tb isolates.
Different mechanisms contribute to drug resistance acquisition. Intrinsic mechanisms developed by M.tb throughout its evolution, such as cell wall permeability, efflux pumps and alteration and degradation of the drug, facilitate the effect of the drug to be neutralised [Reference Nasiri27]. Furthermore, mutations in intergenic regions, such as ahpC-oxyR in the INH resistance, may cause phenotypic resistance without the presence of commonly known mutations [Reference Seifert23].
Our cumulative mutation rate in katG and inhA was 85.5%, a global review carried out by Seifert et al. [Reference Seifert23], which described a similar rate in several studies, affirms that at least 84% of M.tb isolates with INH phenotype resistance are detectable with molecular diagnosis, based on the analysis of these mutations. Besides the fact that these mutations are present in <0.1% of isolates susceptible to INH, the diagnosis through these mutations has a specificity above 99% as markers of phenotypic resistance to INH [Reference Seifert23].
In the analysis of the rpoB gene, the most frequent mutations found were in the 531, 516 and 526 codons, the same as reported in other studies with RIF-resistant isolates [Reference Esteves22, Reference Isakova28]. The mutation in the codon 531 stands out with higher frequency (61.8%), also found in other studies in the same state [Reference Esteves22, Reference Perizzolo29], in other regions of Brazil [Reference De Freitas30, Reference Prim31] and other countries [Reference Isakova28]. The codon 516 in the rpoB gene was mutated in 16.8% of the isolates and 2.3% of them had single nucleotide exchange, other studies show a bigger mutation rate with SNP in this codon [Reference De Freitas30, Reference Hameed32]. The codon 526 was mutated in 9.9% of the isolates, and similar results were found in MDR isolates from a nearby state [Reference Prim31]. Moreover, we found 15 (11.4%) isolates WT for the rpoB gene, similar to that described by Perizzolo et al. [Reference Perizzolo29] and De Freitas et al. [Reference De Freitas30] with samples from the same State.
Another important finding in codon 516 of rpoB was an insertion of 12 nucleotides (CCAGAACAACCC) that was described for the first time by Perizzolo et al. [Reference Perizzolo29]. It was observed in 19/131 (14.5%) of our samples, and a similar frequency was also found by Esteves et al. [Reference Esteves22] in a similar study in the same region of Brazil. According to Esteves et al. [Reference Esteves22], the insertion was present only in isolates classified as LAM family, previously wrongly classified as PINI2 by spoligotyping. However, in this study, we found the insertion present mostly in LAM family (12/19), but also in Haarlem family (5/19), West African family (1/19) and another one in TUR family by the combined analysis of MIRU and spoligotyping. Among the isolates with the insertion, there were two clusters with two isolates each that belonged to bigger clusters (LAM and Haarlem family). We noticed that out of the 19 isolates with this insertion, five were prisoners and one was a worker at the prison system; even though it is not so representative, it could be a way of transmission of these strains.
A study in silico that has been carried out by our group [Reference Rossetti33] indicates that this insertion in codon 516, with a duplication of four amino acids, decreases the binding efficiency of RIF and RNA polymerase through a conformational change in a region close to the RIF binding region. Another study done by Malshetty et al. [Reference Malshetty34] with Mycobacterium smegmatis (M. smegmatis) also shows that an insertion is related to RIF resistance due to changes in spatial conformation in the rpoB amino acids. M. smegmatis and M.tb are very similar, and the rpoB gene is highly conserved among them, so mutations in M. smegmatis model have great relevance in M.tb studies [Reference Malshetty34]. There are still few studies describing this 12-nucleotide insertion in the rpoB gene, all that we could find until now were two studies with sampling originating from the same state of our study; therefore, more studies on this are necessary to clarify its importance in the acquisition and transmission of MDR isolates.
The deletion RDRio was present in 32% of our isolates (42/131), and a similar frequency (38%) was found in the same State [Reference Dalla Costa35] and other regions of Brazil [Reference Barbosa Cde36]. In disagreement with the previous results that found RDRio exclusively in LAM Family [Reference Lazzarini21, Reference Barbosa Cde36, Reference Weisenberg37], we found the deletion also in TUR and Uganda Family in the combined analysis of MIRU-VNTR and spoligotyping, and classified as LAM and T family, respectively, through spoligotyping alone. This finding is contrary to the initial hypothesis of Lazzarini et al. [Reference Lazzarini21] in which the RDRio genotype would be a marker exclusive to LAM sublineage. However, TUR and Uganda family can be a result of convergent evolution and homoplasy, since the molecular markers used in these methodologies can change and emerge into different strains [Reference Allix-Béguec6, Reference Coscolla and Gagneux9, Reference Brudey38, Reference Filliol, Motiwala and Cavatore39].
Some studies associate RDRio deletion with an increased transmissibility of the bacillus [Reference Yoder, Lamichhane and Bishai40], treatment failure [Reference Chatterjee41], drug resistance [Reference Kempker42] and increased number of pulmonary cavitation [Reference Lazzarini43], associating RDRio with a more severe form of TB. However, there are studies that did not find associations between RDRio strains and unfavourable clinical outcome [Reference Barbosa Cde36]. Therefore, further investigation is necessary to understand the relationship between the clinical complications and the RDRio genotype, mainly through X-ray analysis, qualitative clinical data and patient follow-ups.
In the present study, the combined genotype methods (MIRU and spoligotyping) presented LAM family as 65.6% and Haarlem family 22.1% of our isolates, in accordance with other studies in the same state [Reference Esteves22, Reference Perizzolo29, Reference Dalla Costa35] and other regions of Brazil [Reference De Freitas30, Reference Perdigão44]. Most of these studies also show T family as one of the most frequent among Brazilian M.tb strains. In our analysis by spoligotyping alone, we also observed T family as the second most frequent (17.5%), but when combined with MIRU-VNTR, these families were mostly classified as Haarlem and LAM lineage, increasing the number of these genotypes. The same happens observing the isolates classified as PINI2 family by spoligotyping, but identified as LAM genotype in the combined analysis, confirming the study of Dalla Costa et al. [Reference Dalla Costa45]. Differences in classification through MIRU-VNTR and spoligotyping were first described in isolates from Brazil by Vasconcellos et al. [Reference Vasconcellos46] and are in accordance with our study.
The higher frequency of LAM family in the Brazilian territory can be explained by the Portuguese colonisation in the 15th and 16th centuries, since Portugal also shows high frequency of LAM isolates [Reference David47], as well as in African countries such as Mozambique, which also was involved in migratory flow to Brazil in the 16th century [Reference Viegas48]. Different M.tb lineages are associated with specific regions, different human population and different ethnic groups [Reference Brudey38, Reference Gagneux49], thus the human migration process contributed on shaping the current scenario of M.tb population globally [Reference Wirth50].
Some isolates were classified as unknown by MIRU method alone; however, we obtained an identification when we analysed it together with spoligotyping. Regarding the clustering rate, the MIRU-VNTR 24loci rate was 0.13, while spoligotyping was 0.61, showing the importance of the combined analysis for a better classification of the isolates.
About the clustering analysis, 118 M.tb isolates had unique patterns and 13 were grouped in four clusters. Out of these clusters, three had three isolates each and one cluster had four isolates. It was also noticed that in almost every cluster, there were isolates with the 12 nucleotide insertion in the rpoB gene. The presence of clusters with identical genotypes among the isolates is an indicator of the transmission of the same strain [Reference Vluggen51]. In this study, no correlation about the clusters and patient's information was found to determine any kind of transmission, probably due to low clustering rate of the isolates. Our low recent transmission rate (6.8%), determined by the clusters in relation to the total number of isolates, is possibly due to the short time of sampling, same limitation found by Xu et al. [Reference Xu52]. The comparison of recent transmission rate is a valuable information to monitor strain transmission over the years [Reference Hamblion53].
By analysing the patient characteristics of this study, we observed that the majority of the patients was self-employed/salaried employee followed by prisoners and unemployed, with years of schooling from 4 to 7, who had previous treatment failure, HIV co-infection, besides other comorbidities, such as tobacco smoking and consumption of illicit drugs. These show the characteristics of a vulnerable population with a weakened immune system, which favours the acquisition of MDR-TB [Reference Borrell and Gagneux54]. Although studies show that some families are related to some worsening in TB treatment and transmission [Reference Coscolla and Gagneux55, Reference Chae and Shin56], in this study no correlations were observed regarding the patients' characteristics and a specific family, or regarding treatment outcomes and the families.
This study contributes valuable information about the molecular characterisation of MDR strains of the Southern Brazilian population. It helps to understand the transmission dynamics of the period, as well as collaborates with epidemiological data of the most frequent genotypes in the State of RS, thus contributing with the monitoring of MDR-TB strains in the region and the Country.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268819001006.
Author ORCIDs
R. S. Salvato, 0000-0002-5701-9437; R. B. Barcellos, 0000-0003-0901-2422; S. S. Miranda, 0000-0001-7245-4472.
Acknowledgements
The authors would like to thank the National Council of Research – CNPq/MCT – process CNPq/INCT-TB 573548/2008-0 and process: 441499/2014-7 MCTI/CNPQ/Universal 14/2014, the staff from the Laboratory of Bacteriology of Tuberculosis from IPB/LACEN–(CEVS/SES/RS) for helping with processing the samples and culture management and CEVS/SES/RS for the financial support and infrastructure. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.





 Open access
Open access